Emelianova Alina, Garcia Pablo L, Tan Daniel, Joseph Jerelle A
Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, United States.
Omenn-Darling Bioengineering Institute, Princeton University, Princeton, New Jersey 08544, United States.
JACS Au. 2025 Jul 3;5(7):3125-3139. doi: 10.1021/jacsau.5c00291. eCollection 2025 Jul 28.
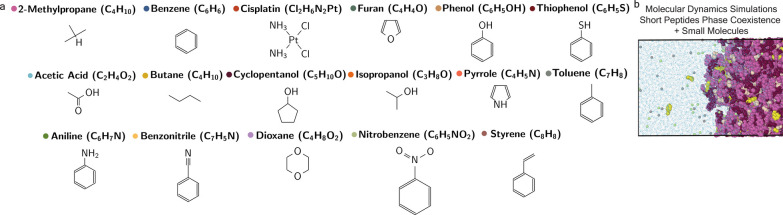
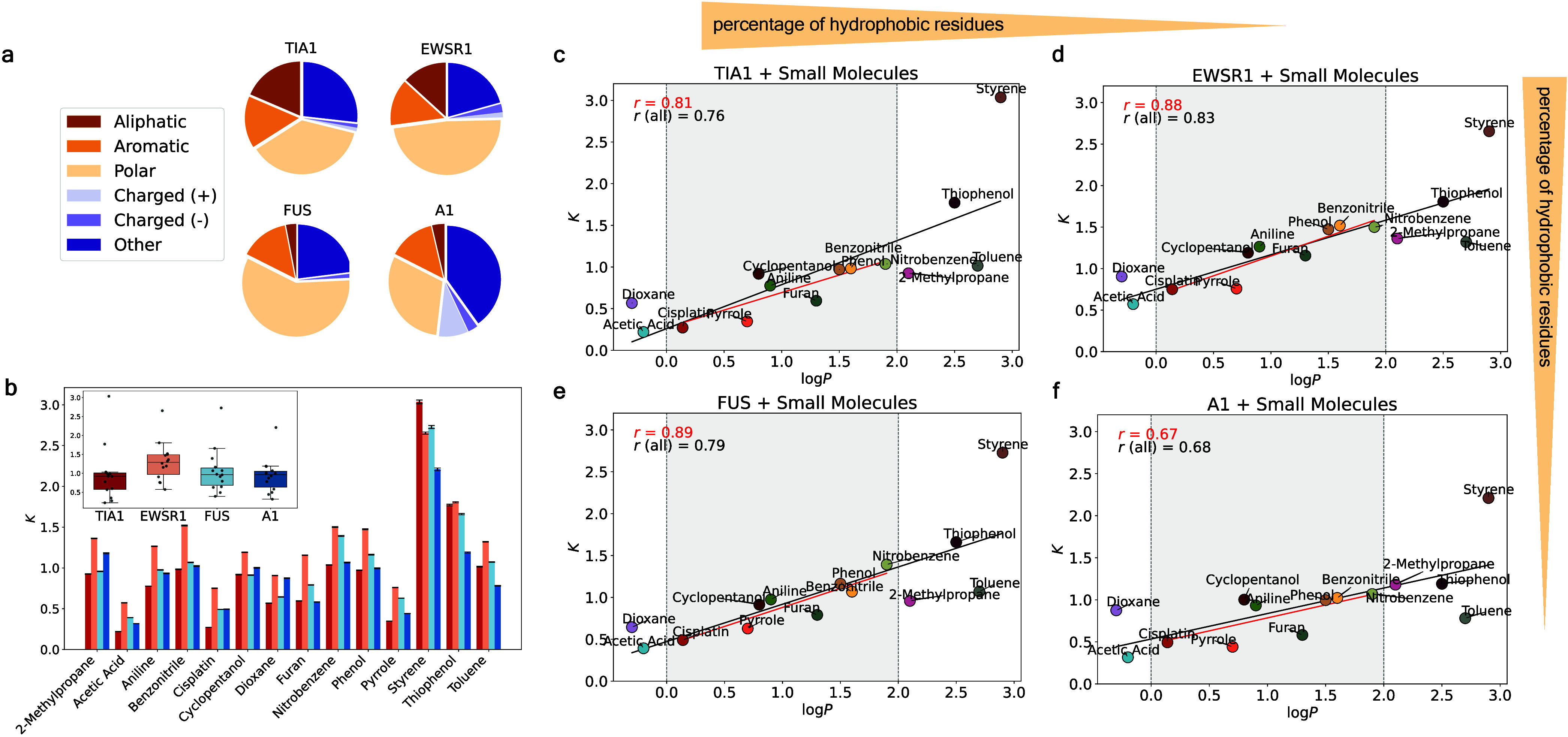
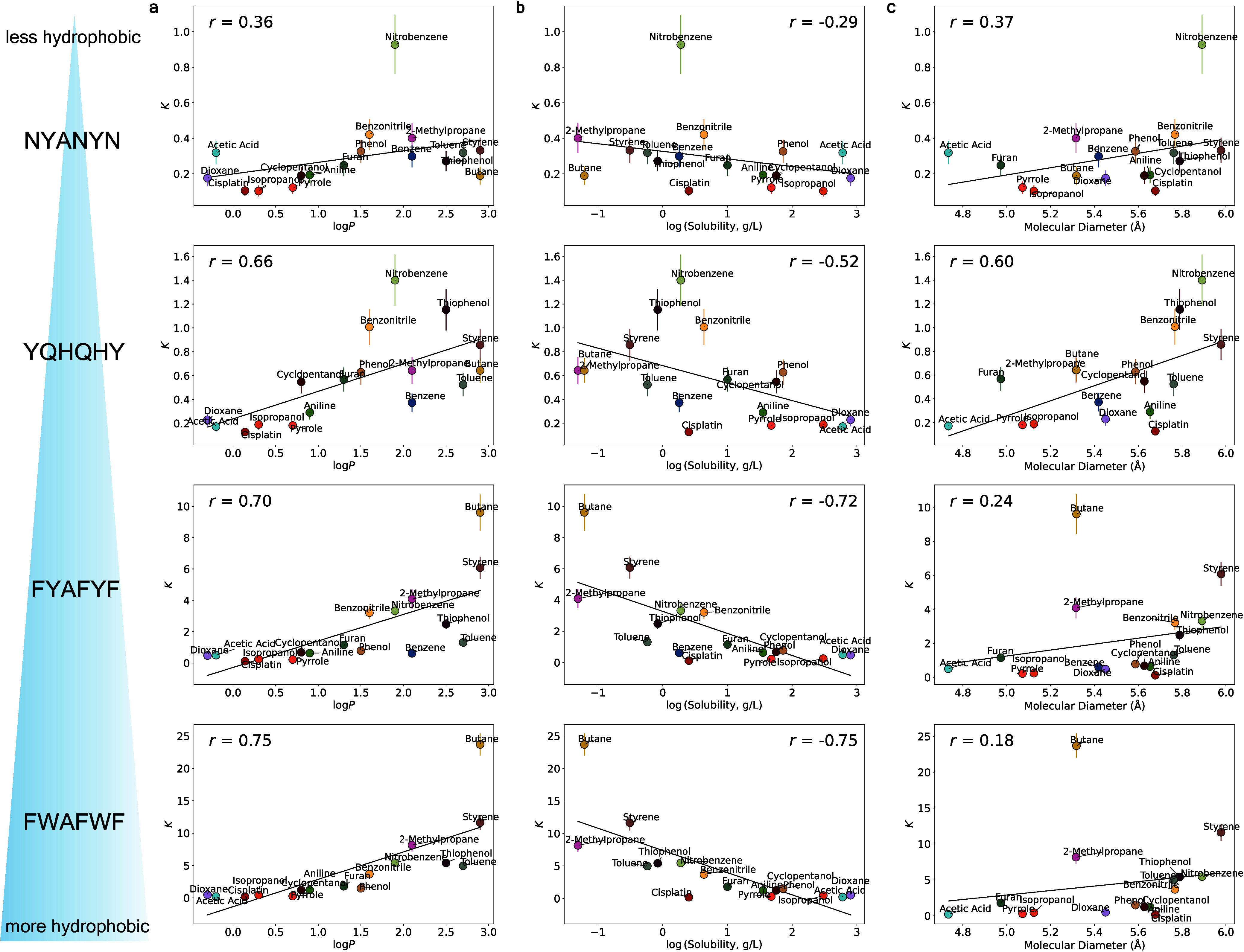
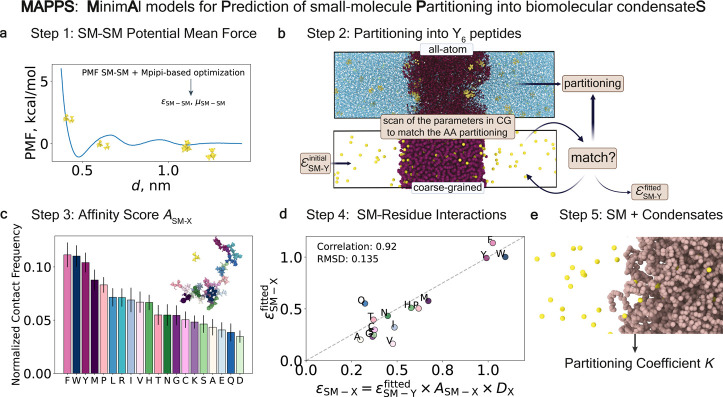
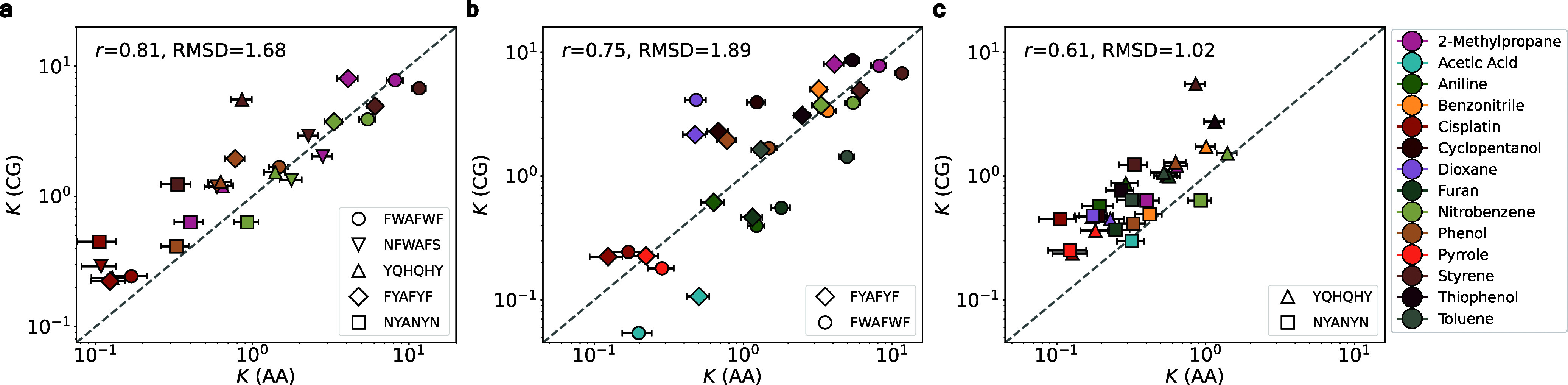
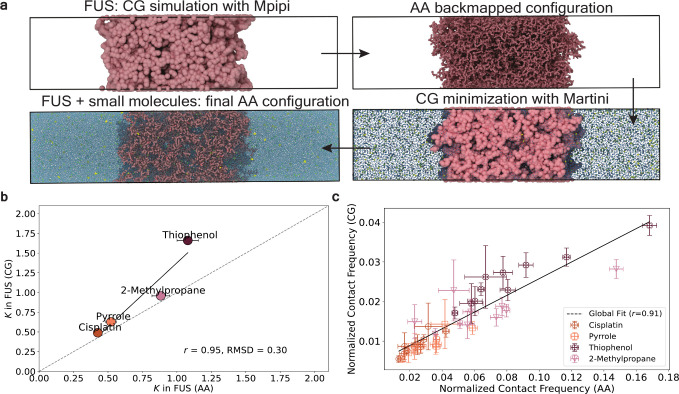
Predicting small-molecule partitioning into biomolecular condensates is the key to developing drugs that selectively target aberrant condensates. However, the molecular mechanisms underlying small-molecule partitioning remain largely unknown. Here, we first exploit atomistic molecular dynamics simulations of model condensates to elucidate the physicochemical rules governing small-molecule partitioning. We find that while hydrophobicity is a key factor in determining partitioning into condensates enriched in hydrophobic residues, partitioning into more polar condensates is driven by specific interactions that can offset the associated entropic cost of localization. The observed selectivity of condensates toward certain compounds suggests that condensate-specific therapeutics can be engineered. Building on these insights, we develop minimal models (MAPPS) for the efficient prediction of small-molecule partitioning into biologically relevant condensates. We demonstrate that this approach reproduces atomistic partition coefficients in both model systems and condensates composed of the low-complexity domain (LCD) of FUS. Applying MAPPS to various LCD-based condensates shows that the protein sequence can exert a selective pressure, thereby influencing small-molecule partitioning. Collectively, our findings reveal that partitioning is driven by both small molecule-protein affinity and the complex interplay between the physicochemical properties of the compounds and the condensate environment.
预测小分子在生物分子凝聚物中的分配是开发选择性靶向异常凝聚物的药物的关键。然而,小分子分配背后的分子机制在很大程度上仍然未知。在这里,我们首先利用模型凝聚物的原子分子动力学模拟来阐明控制小分子分配的物理化学规则。我们发现,虽然疏水性是决定小分子分配到富含疏水残基的凝聚物中的关键因素,但小分子分配到极性更强的凝聚物中是由特定相互作用驱动的,这些相互作用可以抵消相关的定位熵成本。观察到的凝聚物对某些化合物的选择性表明,可以设计出针对特定凝聚物的疗法。基于这些见解,我们开发了最小模型(MAPPS),用于有效预测小分子在生物相关凝聚物中的分配。我们证明,这种方法在模型系统和由FUS的低复杂性结构域(LCD)组成的凝聚物中都能重现原子分配系数。将MAPPS应用于各种基于LCD的凝聚物表明,蛋白质序列可以施加选择性压力,从而影响小分子分配。总的来说,我们的研究结果表明,分配是由小分子与蛋白质的亲和力以及化合物的物理化学性质与凝聚物环境之间的复杂相互作用驱动的。