Lentsch A B, Czermak B J, Bless N M, Ward P A
Department of Pathology, University of Michigan Medical School, Ann Arbor 48109-0602, USA.
Am J Pathol. 1998 May;152(5):1327-36.
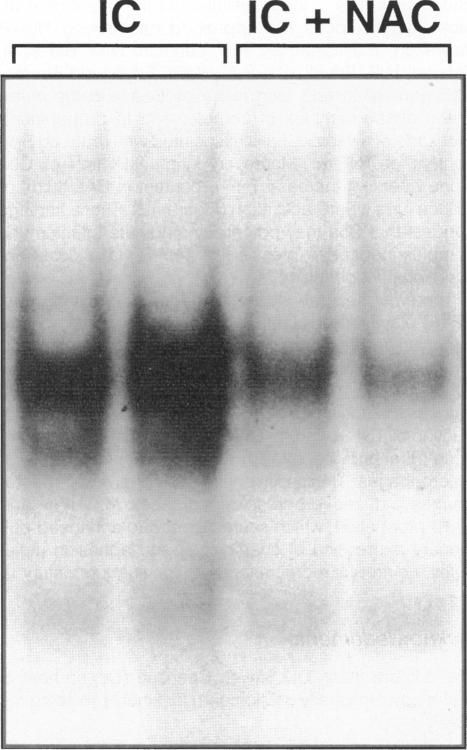
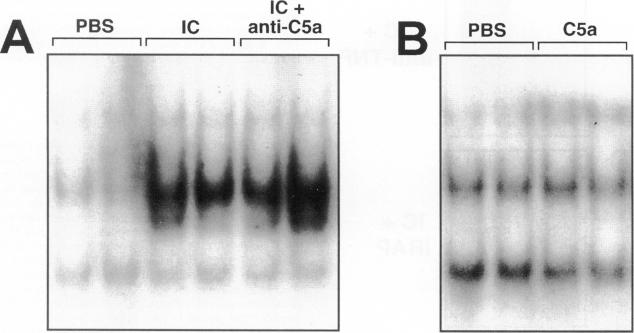
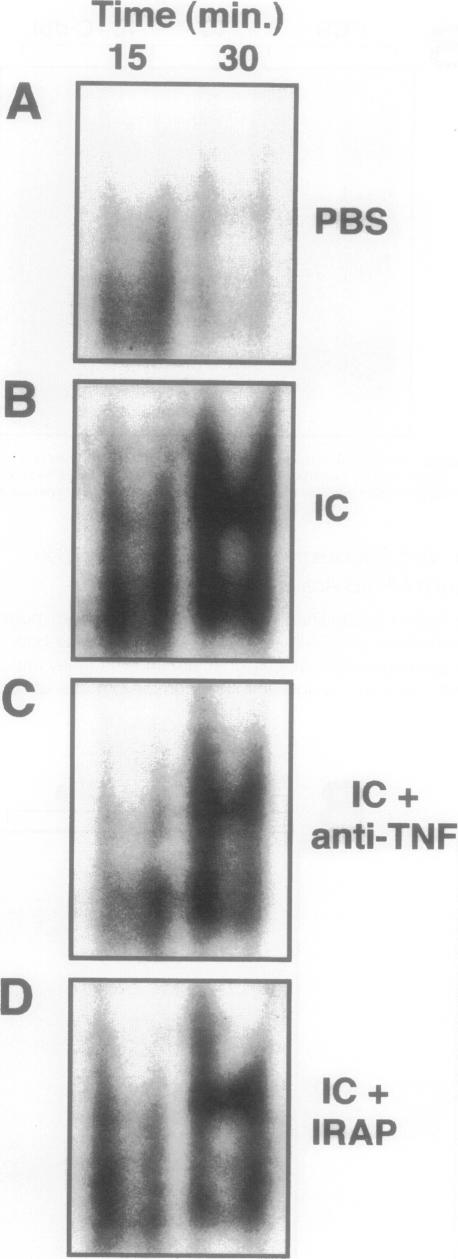
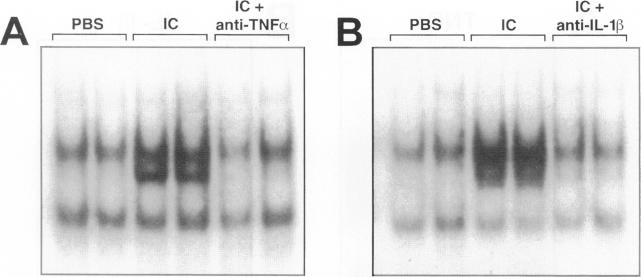
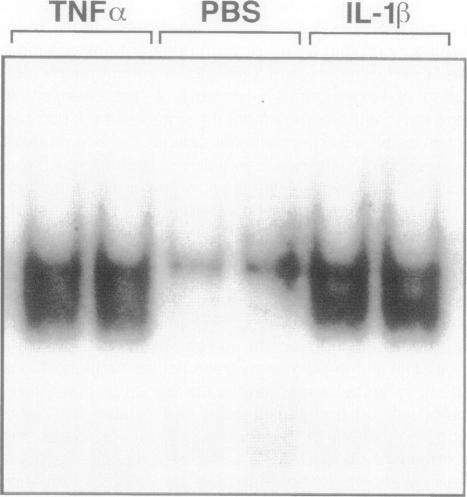
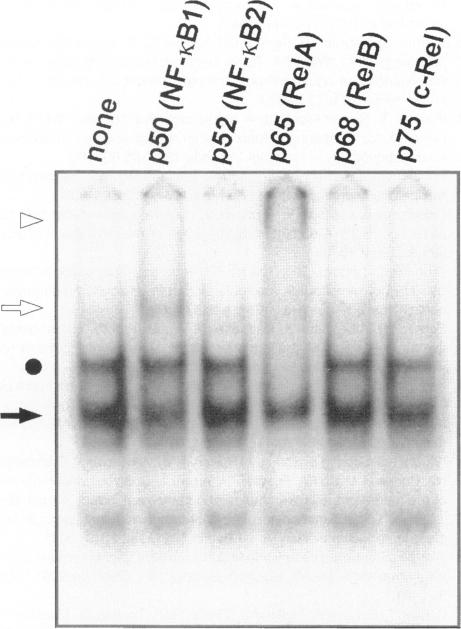
The development of acute lung inflammatory injury induced by alveolar deposition of IgG immune complexes in rats requires increased production of the proinflammatory cytokines, tumor necrosis factor-alpha (TNF-alpha), and interleukin-1beta (IL-1beta) as well as the complement activation product, C5a. Transcription of TNF-alpha and IL-1beta genes are known to be regulated by the nuclear factor-kappa B (NF-kappaB). During IgG immune complex-induced lung inflammation, NF-kappaB has been shown to be activated in both alveolar macrophages and whole lung tissues. In the current studies we sought to determine whether TNF-alpha, IL-1beta, the complement system and oxidants contribute to the activation of NF-kappaB in the lung. Electrophoretic mobility shift analysis of nuclear extracts from whole lung tissues demonstrated that NF-kappaB activation induced by the presence of IgG immune complexes occurred independently of the complement system and neutrophils. Intrapulmonary instillation of TNF-alpha or IL-1beta into normal lung induced NF-kappaB, whereas C5a was incapable of causing NF-kappaB activation. In alveolar macrophages stimulated in vitro with IgG immune complexes, NF-kappaB activation was greatly attenuated in the presence of antibodies to TNF-alpha or IL-1beta. Similarly, in vivo blockade of TNF-alpha or IL-1beta suppressed lung NF-kappaB activation during IgG immune complex-induced lung injury. N-acetylcysteine, but not catalase, suppressed activation of lung NF-kappaB. These data suggest that TNF-alpha and IL-1beta function in an autocrine or paracrine manner to amplify the lung inflammatory response through activation of NF-kappaB. Oxidants not derived from neutrophils also appear to play a role in this process, whereas complement activation products are not involved in this phenomenon.
大鼠肺泡内IgG免疫复合物沉积诱导的急性肺炎症损伤的发展需要促炎细胞因子肿瘤坏死因子-α(TNF-α)和白细胞介素-1β(IL-1β)以及补体激活产物C5a的产生增加。已知TNF-α和IL-1β基因的转录受核因子-κB(NF-κB)调控。在IgG免疫复合物诱导的肺部炎症过程中,NF-κB已被证明在肺泡巨噬细胞和全肺组织中均被激活。在当前研究中,我们试图确定TNF-α、IL-1β、补体系统和氧化剂是否有助于肺部NF-κB的激活。对全肺组织核提取物的电泳迁移率变动分析表明,IgG免疫复合物存在诱导的NF-κB激活独立于补体系统和中性粒细胞。向正常肺内滴注TNF-α或IL-1β可诱导NF-κB,而C5a不能引起NF-κB激活。在体外被IgG免疫复合物刺激的肺泡巨噬细胞中,在存在抗TNF-α或抗IL-1β抗体的情况下,NF-κB激活被大大减弱。同样,在体内阻断TNF-α或IL-1β可抑制IgG免疫复合物诱导的肺损伤期间肺NF-κB的激活。N-乙酰半胱氨酸而非过氧化氢酶可抑制肺NF-κB的激活。这些数据表明,TNF-α和IL-1β以自分泌或旁分泌方式发挥作用,通过激活NF-κB来放大肺部炎症反应。并非源自中性粒细胞的氧化剂似乎也在此过程中起作用,而补体激活产物不参与此现象。