Steffen Martin, Petti Allegra, Aach John, D'haeseleer Patrik, Church George
Dept of Genetics, Harvard Medical School, 200 Longwood Avenue, Boston, Massachusetts, 02115, USA.
BMC Bioinformatics. 2002 Nov 1;3:34. doi: 10.1186/1471-2105-3-34.
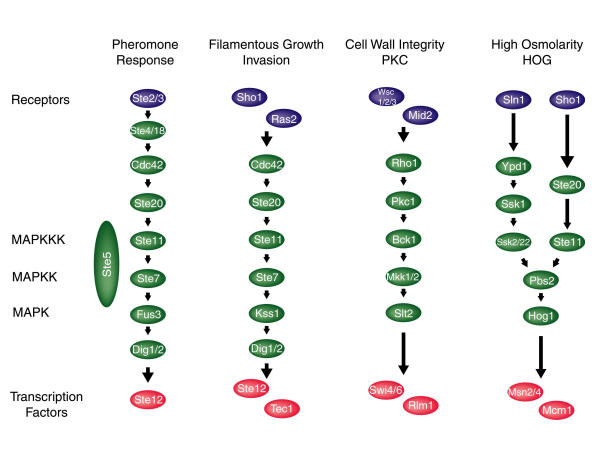
Intracellular signal transduction is achieved by networks of proteins and small molecules that transmit information from the cell surface to the nucleus, where they ultimately effect transcriptional changes. Understanding the mechanisms cells use to accomplish this important process requires a detailed molecular description of the networks involved.
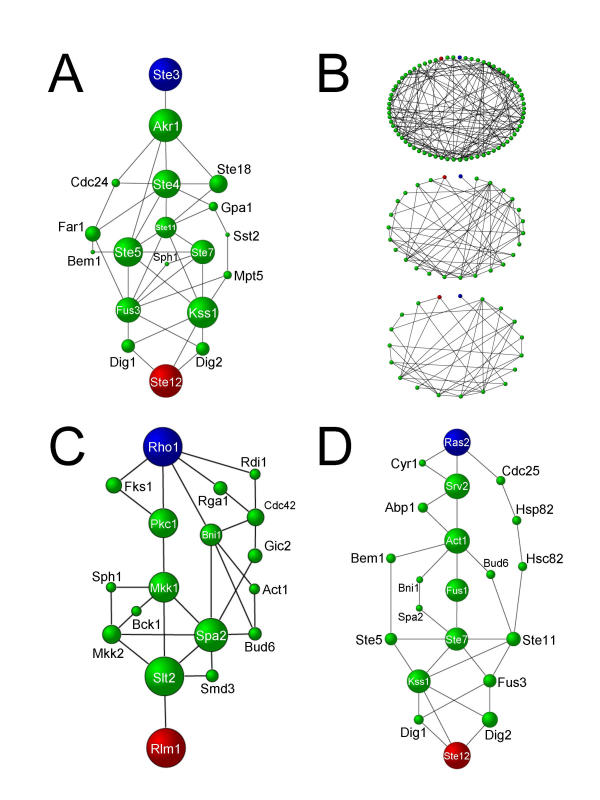
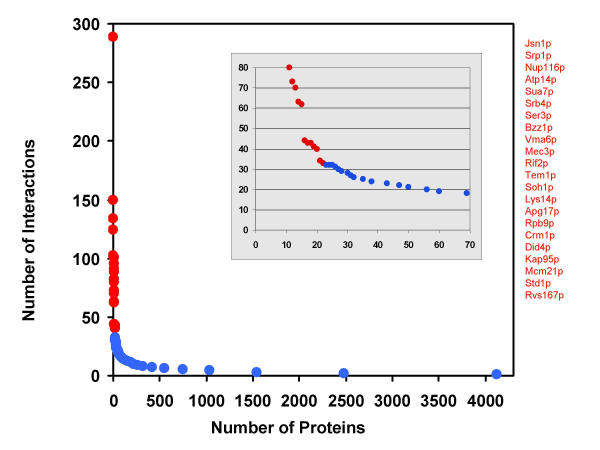
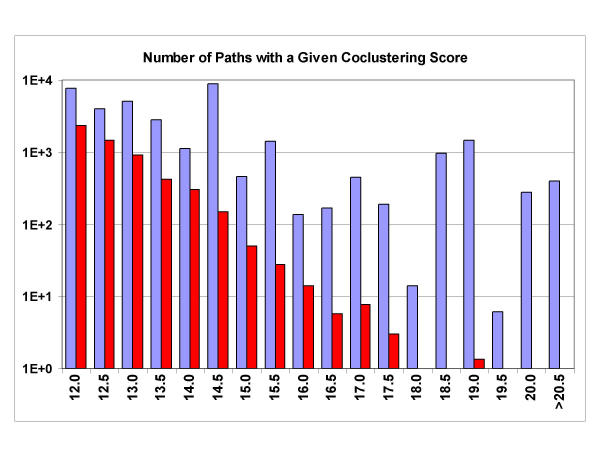
We have developed a computational approach for generating static models of signal transduction networks which utilizes protein-interaction maps generated from large-scale two-hybrid screens and expression profiles from DNA microarrays. Networks are determined entirely by integrating protein-protein interaction data with microarray expression data, without prior knowledge of any pathway intermediates. In effect, this is equivalent to extracting subnetworks of the protein interaction dataset whose members have the most correlated expression profiles.
We show that our technique accurately reconstructs MAP Kinase signaling networks in Saccharomyces cerevisiae. This approach should enhance our ability to model signaling networks and to discover new components of known networks. More generally, it provides a method for synthesizing molecular data, either individual transcript abundance measurements or pairwise protein interactions, into higher level structures, such as pathways and networks.
细胞内信号转导是通过蛋白质和小分子网络实现的,这些网络将信息从细胞表面传递到细胞核,最终在细胞核中影响转录变化。要理解细胞用于完成这一重要过程的机制,需要对所涉及的网络进行详细的分子描述。
我们开发了一种用于生成信号转导网络静态模型的计算方法,该方法利用从大规模双杂交筛选生成的蛋白质相互作用图谱和DNA微阵列的表达谱。网络完全由蛋白质-蛋白质相互作用数据与微阵列表达数据整合确定,无需任何途径中间体的先验知识。实际上,这等同于从蛋白质相互作用数据集中提取其成员具有最相关表达谱的子网。
我们表明我们的技术能够准确重建酿酒酵母中的丝裂原活化蛋白激酶信号网络。这种方法应能增强我们对信号网络进行建模以及发现已知网络新组件的能力。更一般地说,它提供了一种将分子数据(无论是单个转录本丰度测量还是成对蛋白质相互作用)合成到更高层次结构(如途径和网络)中的方法。