Lenhard Boris, Sandelin Albin, Mendoza Luis, Engström Pär, Jareborg Niclas, Wasserman Wyeth W
Center for Genomics and Bioinformatics, Karolinska Institutet, 171 77 Stockholm, Sweden.
Current address: Serono Research and Development, CH-1121 Geneva 20, Switzerland.
J Biol. 2003;2(2):13. doi: 10.1186/1475-4924-2-13. Epub 2003 May 22.
For genes that have been successfully delineated within the human genome sequence, most regulatory sequences remain to be elucidated. The annotation and interpretation process requires additional data resources and significant improvements in computational methods for the detection of regulatory regions. One approach of growing popularity is based on the preferential conservation of functional sequences over the course of evolution by selective pressure, termed 'phylogenetic footprinting'. Mutations are more likely to be disruptive if they appear in functional sites, resulting in a measurable difference in evolution rates between functional and non-functional genomic segments.
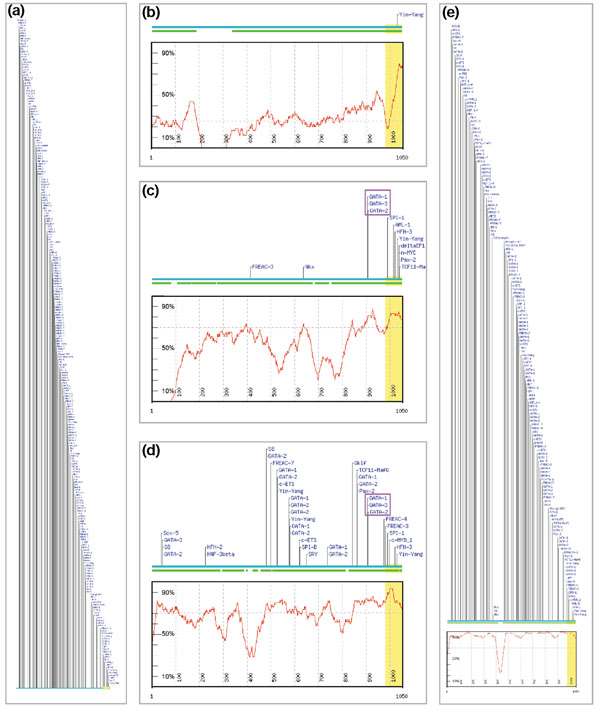
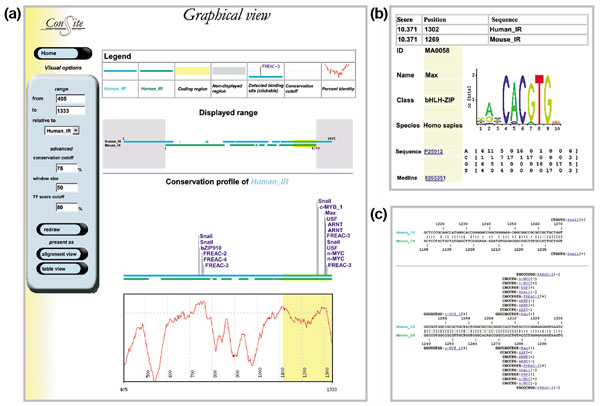
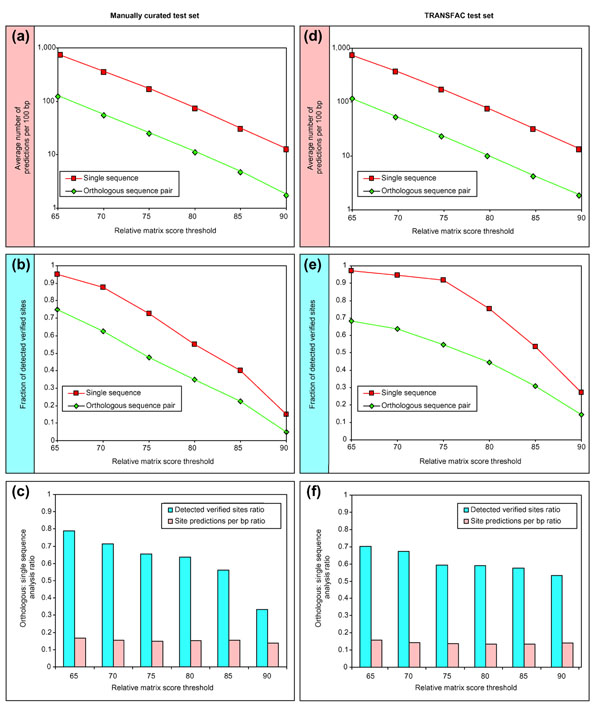
We have devised a flexible suite of methods for the identification and visualization of conserved transcription-factor-binding sites. The system reports those putative transcription-factor-binding sites that are both situated in conserved regions and located as pairs of sites in equivalent positions in alignments between two orthologous sequences. An underlying collection of metazoan transcription-factor-binding profiles was assembled to facilitate the study. This approach results in a significant improvement in the detection of transcription-factor-binding sites because of an increased signal-to-noise ratio, as demonstrated with two sets of promoter sequences. The method is implemented as a graphical web application, ConSite, which is at the disposal of the scientific community at http://www.phylofoot.org/.
Phylogenetic footprinting dramatically improves the predictive selectivity of bioinformatic approaches to the analysis of promoter sequences. ConSite delivers unparalleled performance using a novel database of high-quality binding models for metazoan transcription factors. With a dynamic interface, this bioinformatics tool provides broad access to promoter analysis with phylogenetic footprinting.
对于已在人类基因组序列中成功描绘出的基因,大多数调控序列仍有待阐明。注释和解读过程需要额外的数据资源以及用于检测调控区域的计算方法有显著改进。一种越来越受欢迎的方法是基于功能序列在进化过程中因选择压力而具有的优先保守性,即“系统发育足迹法”。如果突变出现在功能位点,它们更有可能具有破坏性,从而导致功能和非功能基因组片段之间的进化速率存在可测量的差异。
我们设计了一套灵活的方法来识别和可视化保守的转录因子结合位点。该系统报告那些既位于保守区域又在两个直系同源序列比对中的等效位置以位点对形式存在的假定转录因子结合位点。为便于研究,组装了后生动物转录因子结合谱的基础集合。如两组启动子序列所示,由于信噪比提高,这种方法在转录因子结合位点的检测方面有显著改进。该方法以图形化网络应用程序ConSite的形式实现,可在http://www.phylofoot.org/上供科学界使用。
系统发育足迹法极大地提高了生物信息学方法分析启动子序列的预测选择性。ConSite利用后生动物转录因子的高质量结合模型新数据库提供了无与伦比的性能。通过动态界面,这个生物信息学工具为利用系统发育足迹法进行启动子分析提供了广泛的途径。