Wang Weigang, Wyckoff Jeffrey B, Wang Yarong, Bottinger Erwin P, Segall Jeffrey E, Condeelis John S
Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, New York 10461, USA.
BMC Biotechnol. 2003 Aug 12;3:13. doi: 10.1186/1472-6750-3-13.
cDNA microarrays have the potential to identify the genes involved in invasion and metastasis. However, when used with whole tumor tissue, the results average the expression patterns of different cell types. We have combined chemotaxis-based cell collection of the invasive subpopulation of cells within the primary tumor with array-based gene expression analysis to identify the genes necessary for the process of carcinoma cell invasion.
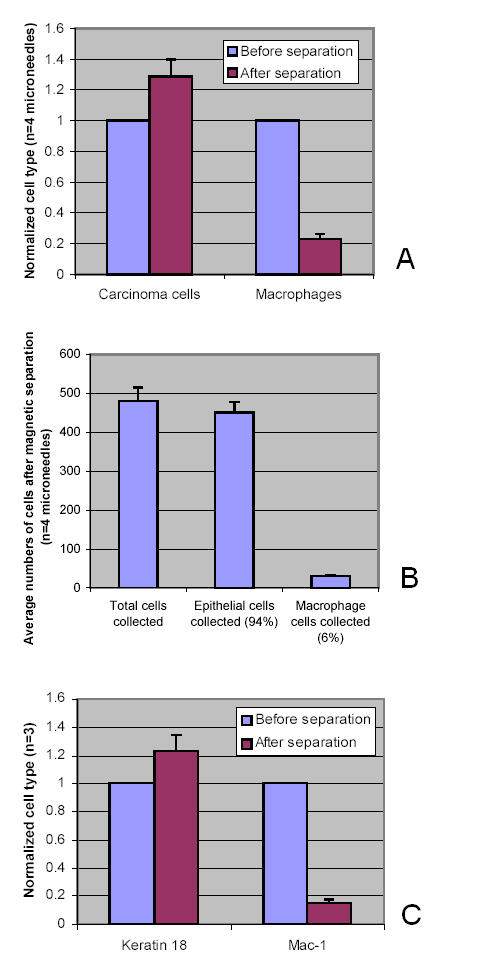
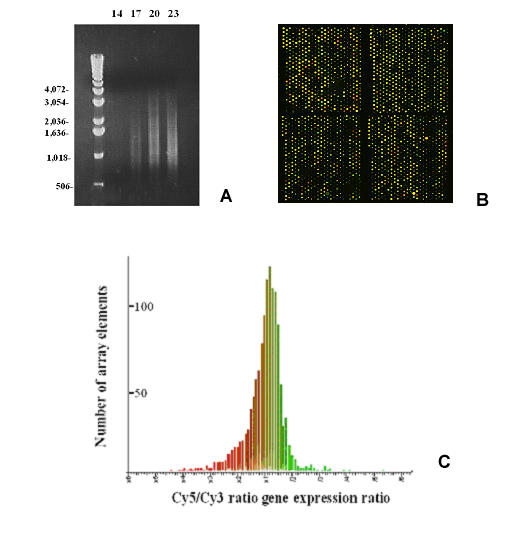
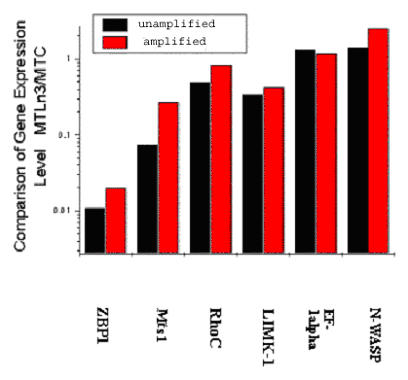
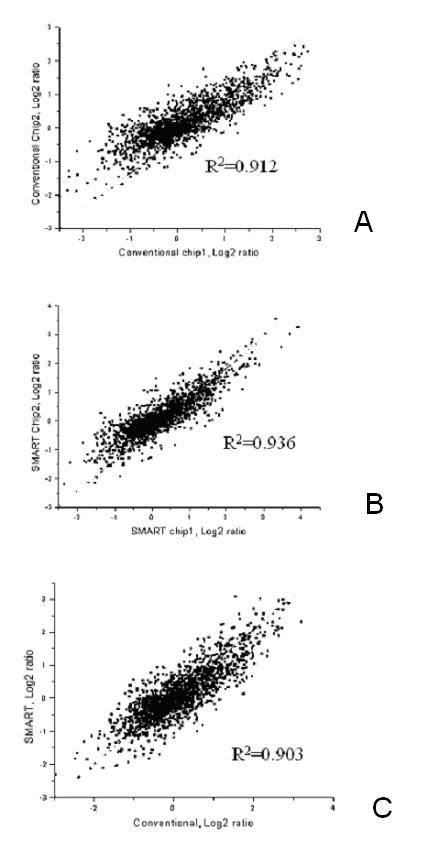
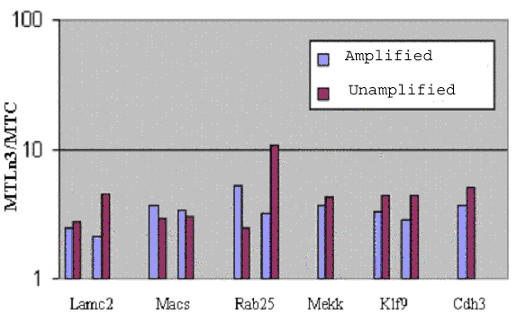
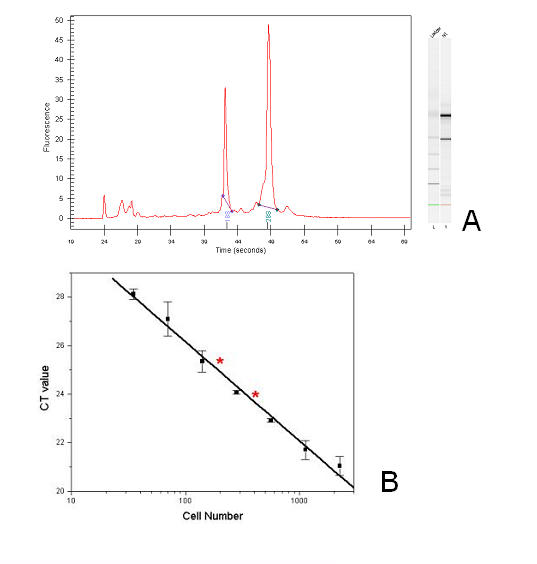
Invasive cells were collected from live primary tumors using microneedles containing chemotactic growth factors to mimic chemotactic signals thought to be present in the primary tumor. When used with mammary tumors of rats and mice, carcinoma cells and macrophages constitute the invasive cell population. Microbeads conjugated with monoclonal anti-CD11b (Mac-1alpha) antibodies were used to separate macrophages from carcinoma cells. We utilized PCR-based cDNA amplification from small number of cells and compared it to the quality and complexity of conventionally generated cDNA to determine if amplified cDNA could be used with fidelity for array analysis of this cell population. These techniques showed a very high level of correlation indicating that the PCR based amplification technique yields a cDNA population that resembles, with high fidelity, the original template population present in the small number of cells used to prepare the cDNA for use with the chip.
The specific collection of invasive cells from a primary tumor and the analysis of gene expression in these cells are is now possible. By further comparing the gene expression patterns of cells collected by invasion into microneedles with that of carcinoma cells obtained from the whole primary tumor, the blood, and whole metastatic tumors, genes that contribute to the invasive process in carcinoma cells may be identified.
cDNA微阵列有潜力识别参与侵袭和转移的基因。然而,当与整个肿瘤组织一起使用时,结果是不同细胞类型表达模式的平均值。我们将基于趋化性的原发性肿瘤内侵袭性细胞亚群的细胞收集与基于阵列的基因表达分析相结合,以识别癌细胞侵袭过程中所需的基因。
使用含有趋化生长因子的微针从活的原发性肿瘤中收集侵袭性细胞,以模拟原发性肿瘤中可能存在的趋化信号。当用于大鼠和小鼠的乳腺肿瘤时,癌细胞和巨噬细胞构成侵袭性细胞群体。用与单克隆抗CD11b(Mac-1α)抗体偶联的微珠将巨噬细胞与癌细胞分离。我们利用基于PCR的少量细胞cDNA扩增,并将其与传统方法产生的cDNA的质量和复杂性进行比较,以确定扩增的cDNA是否可忠实地用于该细胞群体的阵列分析。这些技术显示出非常高的相关性,表明基于PCR的扩增技术产生的cDNA群体与用于制备芯片cDNA的少量细胞中存在的原始模板群体高度相似。
现在可以从原发性肿瘤中特异性收集侵袭性细胞并分析这些细胞中的基因表达。通过进一步比较通过侵入微针收集的细胞与从整个原发性肿瘤、血液和整个转移性肿瘤中获得的癌细胞的基因表达模式,可能识别出有助于癌细胞侵袭过程的基因。