Xue Yu, Zhou Fengfeng, Zhu Minjie, Ahmed Kashif, Chen Guoliang, Yao Xuebiao
School of Life Science, University of Science and Technology of China, Hefei, Anhui 230027, PR China.
Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W184-7. doi: 10.1093/nar/gki393.
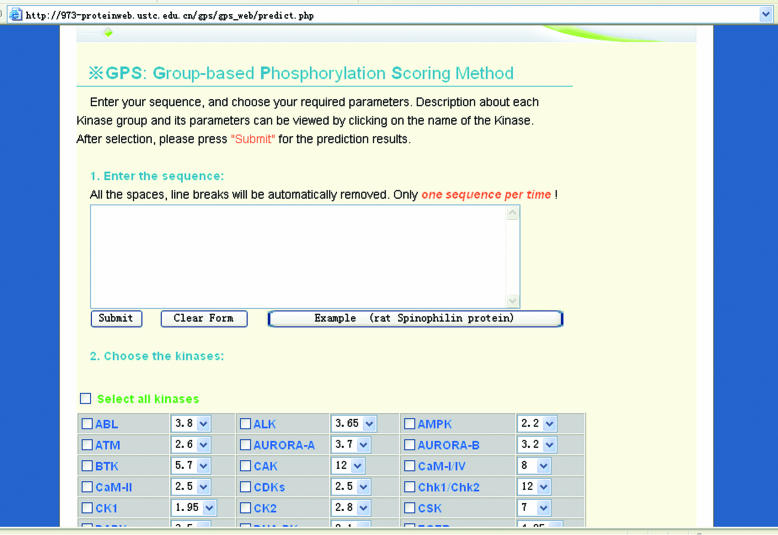
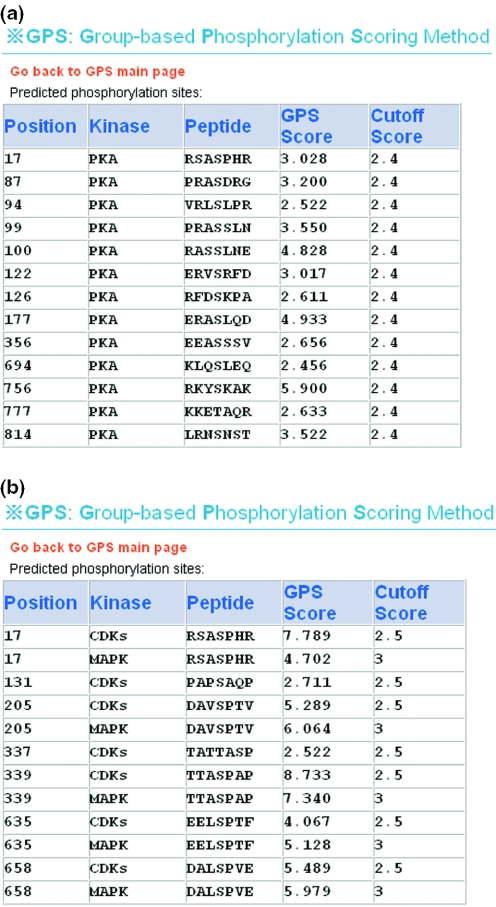
Protein phosphorylation plays a fundamental role in most of the cellular regulatory pathways. Experimental identification of protein kinases' (PKs) substrates with their phosphorylation sites is labor-intensive and often limited by the availability and optimization of enzymatic reactions. Recently, large-scale analysis of the phosphoproteome by the mass spectrometry (MS) has become a popular approach. But experimentally, it is still difficult to distinguish the kinase-specific sites on the substrates. In this regard, the in silico prediction of phosphorylation sites with their specific kinases using protein's primary sequences may provide guidelines for further experimental consideration and interpretation of MS phosphoproteomic data. A variety of such tools exists over the Internet and provides the predictions for at most 30 PK subfamilies. We downloaded the verified phosphorylation sites from the public databases and curated the literature extensively for recently found phosphorylation sites. With the hypothesis that PKs in the same subfamily share similar consensus sequences/motifs/functional patterns on substrates, we clustered the 216 unique PKs in 71 PK groups, according to the BLAST results and protein annotations. Then, we applied the group-based phosphorylation scoring (GPS) method on the data set; here, we present a comprehensive PK-specific prediction server GPS, which could predict kinase-specific phosphorylation sites from protein primary sequences for 71 different PK groups. GPS has been implemented in PHP and is available on a www server at http://973-proteinweb.ustc.edu.cn/gps/gps_web/.
蛋白质磷酸化在大多数细胞调节途径中发挥着重要作用。通过实验鉴定蛋白激酶(PKs)的底物及其磷酸化位点是一项劳动密集型工作,并且常常受到酶促反应可用性和优化的限制。最近,通过质谱(MS)对磷酸化蛋白质组进行大规模分析已成为一种流行的方法。但在实验上,仍然难以区分底物上激酶特异性位点。在这方面,利用蛋白质一级序列对磷酸化位点及其特定激酶进行计算机预测,可为进一步的实验考量和MS磷酸化蛋白质组学数据的解释提供指导。互联网上存在各种此类工具,最多可为30个PK亚家族提供预测。我们从公共数据库下载了经过验证的磷酸化位点,并广泛查阅文献以获取最近发现的磷酸化位点。基于同一亚家族中的PKs在底物上共享相似的共有序列/基序/功能模式这一假设,我们根据BLAST结果和蛋白质注释,将216个独特的PKs聚类为71个PK组。然后,我们在数据集上应用基于组的磷酸化评分(GPS)方法;在此,我们展示了一个全面的PK特异性预测服务器GPS,它可以从蛋白质一级序列预测71个不同PK组的激酶特异性磷酸化位点。GPS已用PHP实现,可在网址为http://973-proteinweb.ustc.edu.cn/gps/gps_web/的万维网服务器上获取。