Naylor Tara L, Greshock Joel, Wang Yan, Colligon Theresa, Yu Q C, Clemmer Virginia, Zaks Tal Z, Weber Barbara L
Abramson Family Cancer Research Institute, University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA.
Breast Cancer Res. 2005;7(6):R1186-98. doi: 10.1186/bcr1356. Epub 2005 Nov 24.
Genomic aberrations in the form of subchromosomal DNA copy number changes are a hallmark of epithelial cancers, including breast cancer. The goal of the present study was to analyze such aberrations in breast cancer at high resolution.
We employed high-resolution array comparative genomic hybridization with 4,134 bacterial artificial chromosomes that cover the genome at 0.9 megabase resolution to analyze 47 primary breast tumors and 18 breast cancer cell lines.
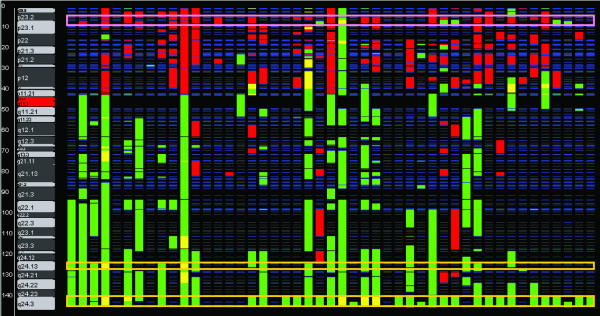
Common amplicons included 8q24.3 (amplified in 79% of tumors, with 5/47 exhibiting high level amplification), 1q32.1 and 16p13.3 (amplified in 66% and 57% of tumors, respectively). Moreover, we found several positive correlations between specific amplicons from different chromosomes, suggesting the existence of cooperating genetic loci. Queried by gene, the most frequently amplified kinase was PTK2 (79% of tumors), whereas the most frequently lost kinase was PTK2B (hemizygous loss in 34% of tumors). Amplification of ERBB2 as measured by comparative genomic hybridization (CGH) correlated closely with ERBB2 DNA and RNA levels measured by quantitative PCR as well as with ERBB2 protein levels. The overall frequency of recurrent losses was lower, with no region lost in more than 50% of tumors; the most frequently lost tumor suppressor gene was RB1 (hemizygous loss in 26% of tumors). Finally, we find that specific copy number changes in cell lines closely mimicked those in primary tumors, with an overall Pearson correlation coefficient of 0.843 for gains and 0.734 for losses.
High resolution CGH analysis of breast cancer reveals several regions where DNA copy number is commonly gained or lost, that non-random correlations between specific amplicons exist, and that specific genetic alterations are maintained in breast cancer cell lines despite repeat passage in tissue culture. These observations suggest that genes within these regions are critical to the malignant phenotype and may thus serve as future therapeutic targets.
亚染色体DNA拷贝数变化形式的基因组畸变是包括乳腺癌在内的上皮癌的一个标志。本研究的目的是在高分辨率下分析乳腺癌中的此类畸变。
我们采用了高分辨率阵列比较基因组杂交技术,使用4134个细菌人工染色体,以0.9兆碱基的分辨率覆盖基因组,分析了47个原发性乳腺肿瘤和18个乳腺癌细胞系。
常见的扩增子包括8q24.3(在79%的肿瘤中扩增,47个中有5个表现出高水平扩增)、1q32.1和16p13.3(分别在66%和57%的肿瘤中扩增)。此外,我们发现来自不同染色体的特定扩增子之间存在若干正相关,表明存在协同作用的基因座。按基因查询,最常扩增的激酶是PTK2(79%的肿瘤),而最常缺失的激酶是PTK2B(34%的肿瘤中半合子缺失)。通过比较基因组杂交(CGH)测量的ERBB2扩增与通过定量PCR测量的ERBB2 DNA和RNA水平以及ERBB2蛋白水平密切相关。复发性缺失的总体频率较低,没有一个区域在超过50%的肿瘤中缺失;最常缺失的肿瘤抑制基因是RB1(26%的肿瘤中半合子缺失)。最后,我们发现细胞系中的特定拷贝数变化与原发性肿瘤中的变化密切相似,增益的总体皮尔逊相关系数为0.843,缺失的为0.734。
乳腺癌的高分辨率CGH分析揭示了几个DNA拷贝数通常增加或减少的区域,特定扩增子之间存在非随机相关性,并且尽管在组织培养中反复传代,乳腺癌细胞系中仍维持特定的基因改变。这些观察结果表明这些区域内的基因对恶性表型至关重要,因此可能作为未来的治疗靶点。