Bailey Timothy L, Williams Nadya, Misleh Chris, Li Wilfred W
Institute of Molecular Bioscience, The University of Queensland, St Lucia, QLD 4072, Australia.
Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W369-73. doi: 10.1093/nar/gkl198.
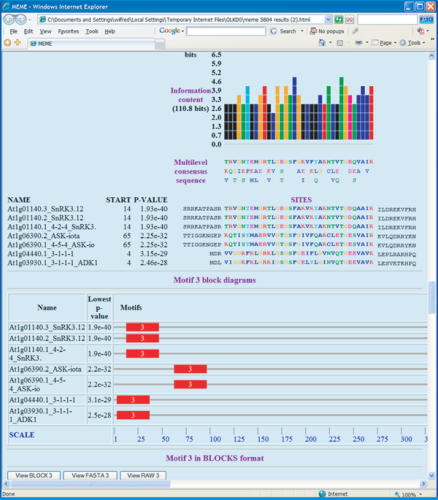
MEME (Multiple EM for Motif Elicitation) is one of the most widely used tools for searching for novel 'signals' in sets of biological sequences. Applications include the discovery of new transcription factor binding sites and protein domains. MEME works by searching for repeated, ungapped sequence patterns that occur in the DNA or protein sequences provided by the user. Users can perform MEME searches via the web server hosted by the National Biomedical Computation Resource (http://meme.nbcr.net) and several mirror sites. Through the same web server, users can also access the Motif Alignment and Search Tool to search sequence databases for matches to motifs encoded in several popular formats. By clicking on buttons in the MEME output, users can compare the motifs discovered in their input sequences with databases of known motifs, search sequence databases for matches to the motifs and display the motifs in various formats. This article describes the freely accessible web server and its architecture, and discusses ways to use MEME effectively to find new sequence patterns in biological sequences and analyze their significance.
MEME(用于基序引出的多重期望最大化算法)是在生物序列集中搜索新型“信号”时使用最广泛的工具之一。其应用包括发现新的转录因子结合位点和蛋白质结构域。MEME通过搜索用户提供的DNA或蛋白质序列中出现的重复、无间隙序列模式来工作。用户可以通过由国家生物医学计算资源中心托管的网络服务器(http://meme.nbcr.net)以及几个镜像站点进行MEME搜索。通过同一个网络服务器,用户还可以访问基序比对和搜索工具,以在序列数据库中搜索与几种流行格式编码的基序相匹配的序列。通过点击MEME输出中的按钮,用户可以将在其输入序列中发现的基序与已知基序数据库进行比较,在序列数据库中搜索与基序相匹配的序列,并以各种格式显示这些基序。本文描述了这个可免费访问的网络服务器及其架构,并讨论了有效使用MEME在生物序列中找到新的序列模式并分析其意义的方法。