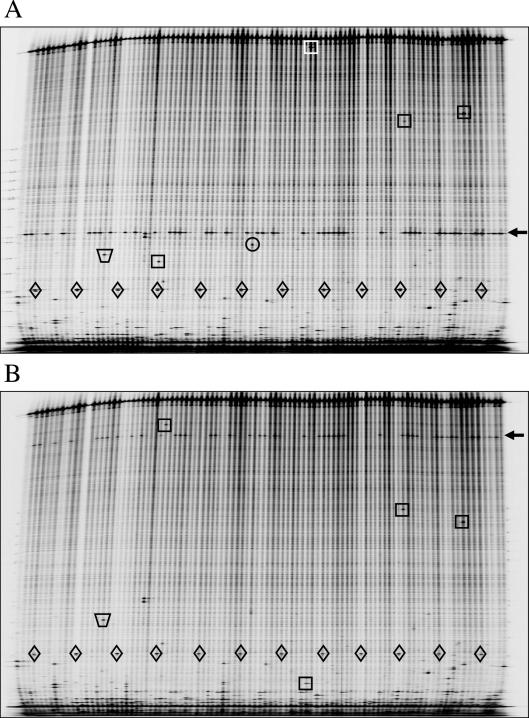
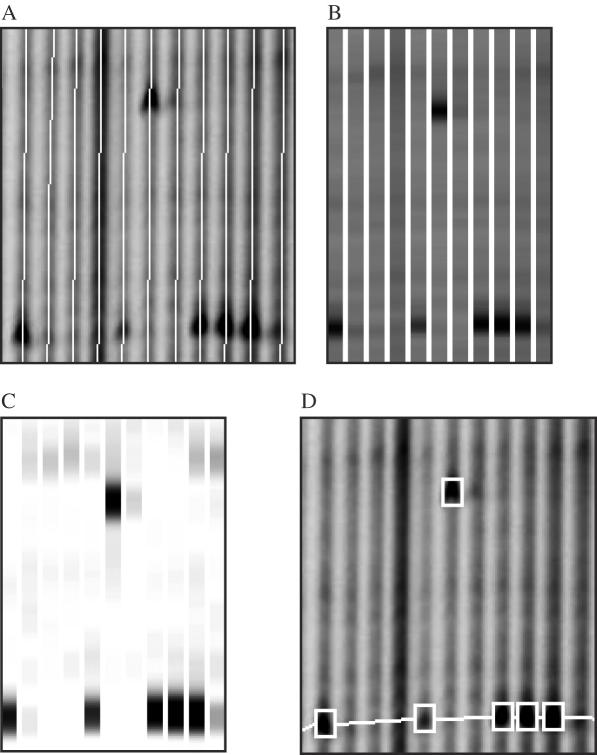
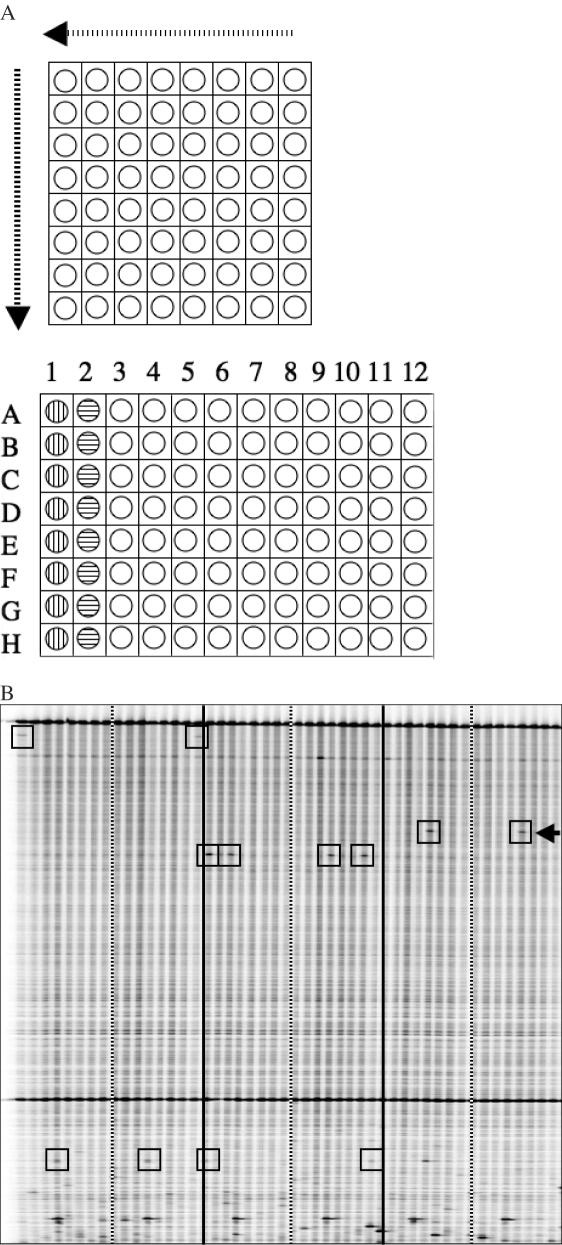
Till Bradley J, Zerr Troy, Bowers Elisabeth, Greene Elizabeth A, Comai Luca, Henikoff Steven
Basic Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Nucleic Acids Res. 2006 Aug 7;34(13):e99. doi: 10.1093/nar/gkl479.
Human individuals differ from one another at only approximately 0.1% of nucleotide positions, but these single nucleotide differences account for most heritable phenotypic variation. Large-scale efforts to discover and genotype human variation have been limited to common polymorphisms. However, these efforts overlook rare nucleotide changes that may contribute to phenotypic diversity and genetic disorders, including cancer. Thus, there is an increasing need for high-throughput methods to robustly detect rare nucleotide differences. Toward this end, we have adapted the mismatch discovery method known as Ecotilling for the discovery of human single nucleotide polymorphisms. To increase throughput and reduce costs, we developed a universal primer strategy and implemented algorithms for automated band detection. Ecotilling was validated by screening 90 human DNA samples for nucleotide changes in 5 gene targets and by comparing results to public resequencing data. To increase throughput for discovery of rare alleles, we pooled samples 8-fold and found Ecotilling to be efficient relative to resequencing, with a false negative rate of 5% and a false discovery rate of 4%. We identified 28 new rare alleles, including some that are predicted to damage protein function. The detection of rare damaging mutations has implications for models of human disease.
人类个体之间在核苷酸位点上仅有约0.1%的差异,但这些单核苷酸差异却构成了大部分可遗传的表型变异。发现人类变异并对其进行基因分型的大规模研究一直局限于常见的多态性。然而,这些研究忽略了可能导致表型多样性和遗传疾病(包括癌症)的罕见核苷酸变化。因此,对高通量方法以可靠检测罕见核苷酸差异的需求日益增加。为此,我们采用了一种名为Ecotilling的错配发现方法来发现人类单核苷酸多态性。为了提高通量并降低成本,我们开发了通用引物策略并实施了自动条带检测算法。通过对90个人类DNA样本进行5个基因靶点的核苷酸变化筛选,并将结果与公开的重测序数据进行比较,验证了Ecotilling方法。为了提高发现罕见等位基因的通量,我们将样本进行8倍混合,发现Ecotilling相对于重测序是有效的,假阴性率为5%,假发现率为4%。我们鉴定出28个新的罕见等位基因,其中一些预计会损害蛋白质功能。罕见有害突变的检测对人类疾病模型具有重要意义。