Katz Simon, Irizarry Rafael A, Lin Xue, Tripputi Mark, Porter Mark W
Gene Logic Inc., 610 Professional Dr, Gaithersburg, MD, 20876, USA.
BMC Bioinformatics. 2006 Oct 23;7:464. doi: 10.1186/1471-2105-7-464.
Many of the most popular pre-processing methods for Affymetrix expression arrays, such as RMA, gcRMA, and PLIER, simultaneously analyze data across a set of predetermined arrays to improve precision of the final measures of expression. One problem associated with these algorithms is that expression measurements for a particular sample are highly dependent on the set of samples used for normalization and results obtained by normalization with a different set may not be comparable. A related problem is that an organization producing and/or storing large amounts of data in a sequential fashion will need to either re-run the pre-processing algorithm every time an array is added or store them in batches that are pre-processed together. Furthermore, pre-processing of large numbers of arrays requires loading all the feature-level data into memory which is a difficult task even with modern computers. We utilize a scheme that produces all the information necessary for pre-processing using a very large training set that can be used for summarization of samples outside of the training set. All subsequent pre-processing tasks can be done on an individual array basis. We demonstrate the utility of this approach by defining a new version of the Robust Multi-chip Averaging (RMA) algorithm which we refer to as refRMA.
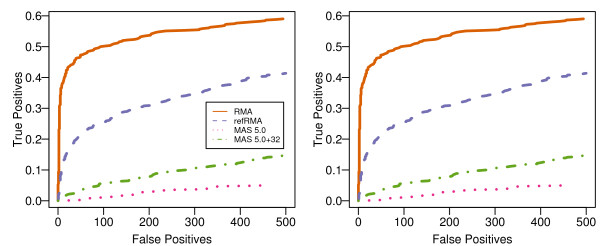
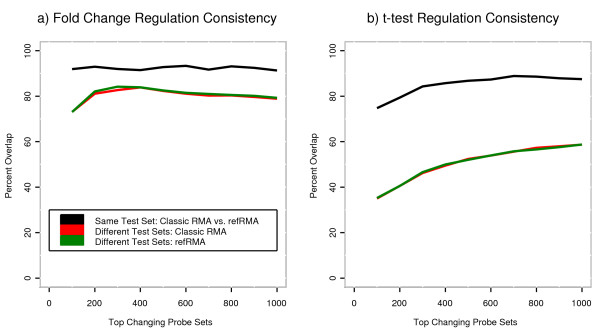
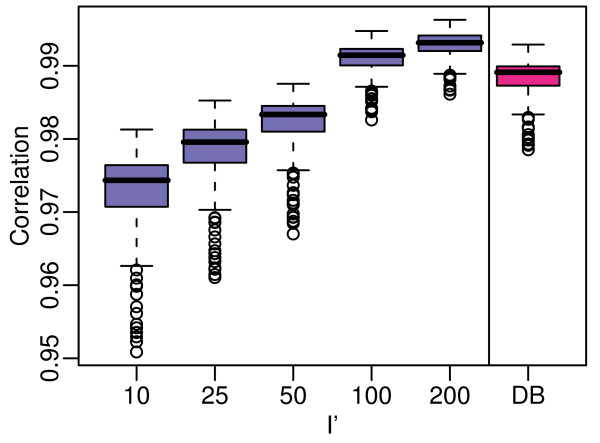
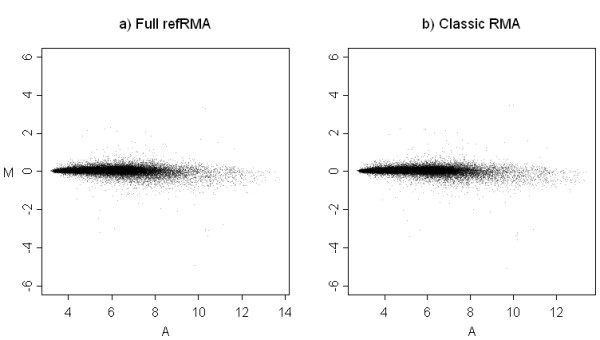
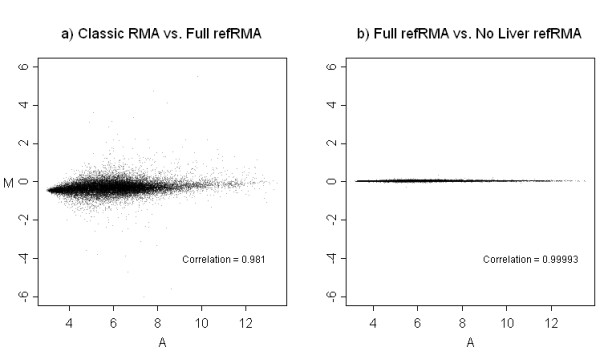
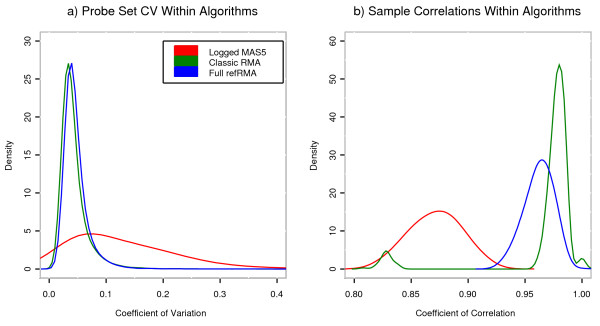
We assess performance based on multiple sets of samples processed over HG U133A Affymetrix GeneChip arrays. We show that the refRMA workflow, when used in conjunction with a large, biologically diverse training set, results in the same general characteristics as that of RMA in its classic form when comparing overall data structure, sample-to-sample correlation, and variation. Further, we demonstrate that the refRMA workflow and reference set can be robustly applied to naïve organ types and to benchmark data where its performance indicates respectable results.
Our results indicate that a biologically diverse reference database can be used to train a model for estimating probe set intensities of exclusive test sets, while retaining the overall characteristics of the base algorithm. Although the results we present are specific for RMA, similar versions of other multi-array normalization and summarization schemes can be developed.
许多用于Affymetrix表达阵列的最流行的预处理方法,如RMA、gcRMA和PLIER,会同时分析一组预定阵列中的数据,以提高最终表达量度的精度。与这些算法相关的一个问题是,特定样本的表达测量高度依赖于用于归一化的样本集,并且用不同的样本集进行归一化得到的结果可能不可比。一个相关问题是,以顺序方式生成和/或存储大量数据的机构,要么每次添加一个阵列时都重新运行预处理算法,要么将它们按批存储并一起进行预处理。此外,对大量阵列进行预处理需要将所有特征级数据加载到内存中,即使使用现代计算机,这也是一项艰巨的任务。我们采用一种方案,该方案使用一个非常大的训练集生成预处理所需的所有信息,该训练集可用于汇总训练集之外的样本。所有后续的预处理任务都可以在单个阵列的基础上完成。我们通过定义一种新的稳健多芯片平均(RMA)算法(我们称之为refRMA)来证明这种方法的实用性。
我们基于在HG U133A Affymetrix基因芯片阵列上处理的多组样本评估性能。我们表明,当refRMA工作流程与一个大型的、生物多样性丰富的训练集结合使用时,在比较整体数据结构、样本间相关性和变异性时,其产生的总体特征与经典形式的RMA相同。此外,我们证明refRMA工作流程和参考集可以稳健地应用于未经处理的器官类型和基准数据,其性能显示出可观的结果。
我们的结果表明,一个生物多样性丰富的参考数据库可用于训练一个模型,以估计排他性测试集的探针集强度,同时保留基础算法的总体特征。尽管我们给出的结果是针对RMA的,但也可以开发其他多阵列归一化和汇总方案的类似版本。