Doytchinova Irini A, Flower Darren R
Faculty of Pharmacy, Medical University of Sofia, 2 Dunav St., 1000 Sofia, Bulgaria.
BMC Bioinformatics. 2007 Jan 5;8:4. doi: 10.1186/1471-2105-8-4.
Vaccine development in the post-genomic era often begins with the in silico screening of genome information, with the most probable protective antigens being predicted rather than requiring causative microorganisms to be grown. Despite the obvious advantages of this approach--such as speed and cost efficiency--its success remains dependent on the accuracy of antigen prediction. Most approaches use sequence alignment to identify antigens. This is problematic for several reasons. Some proteins lack obvious sequence similarity, although they may share similar structures and biological properties. The antigenicity of a sequence may be encoded in a subtle and recondite manner not amendable to direct identification by sequence alignment. The discovery of truly novel antigens will be frustrated by their lack of similarity to antigens of known provenance. To overcome the limitations of alignment-dependent methods, we propose a new alignment-free approach for antigen prediction, which is based on auto cross covariance (ACC) transformation of protein sequences into uniform vectors of principal amino acid properties.
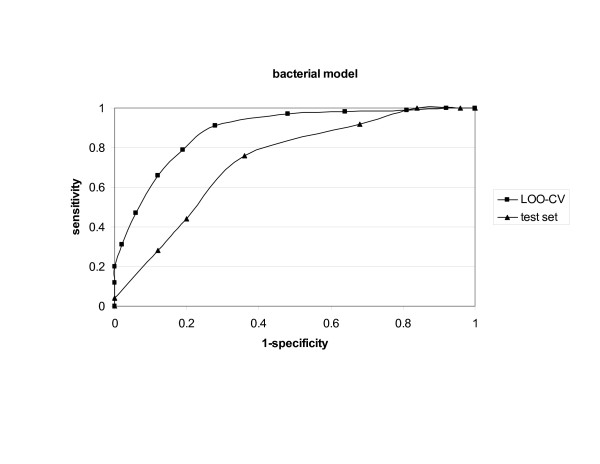
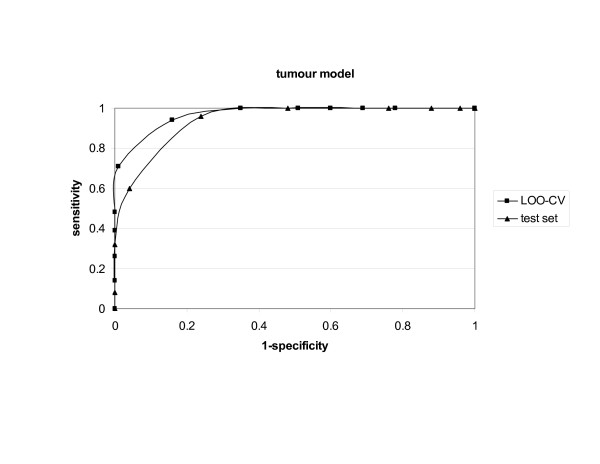
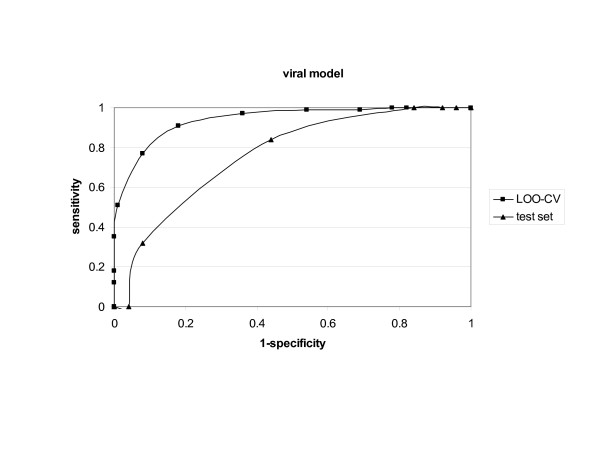
Bacterial, viral and tumour protein datasets were used to derive models for prediction of whole protein antigenicity. Every set consisted of 100 known antigens and 100 non-antigens. The derived models were tested by internal leave-one-out cross-validation and external validation using test sets. An additional five training sets for each class of antigens were used to test the stability of the discrimination between antigens and non-antigens. The models performed well in both validations showing prediction accuracy of 70% to 89%. The models were implemented in a server, which we call VaxiJen.
VaxiJen is the first server for alignment-independent prediction of protective antigens. It was developed to allow antigen classification solely based on the physicochemical properties of proteins without recourse to sequence alignment. The server can be used on its own or in combination with alignment-based prediction methods. It is freely-available online at the URL: http://www.jenner.ac.uk/VaxiJen.
后基因组时代的疫苗开发通常始于对基因组信息的计算机筛选,通过预测最有可能的保护性抗原,而无需培养致病微生物。尽管这种方法具有明显的优势,如速度快和成本效益高,但其成功仍取决于抗原预测的准确性。大多数方法使用序列比对来识别抗原。这存在几个问题。一些蛋白质缺乏明显的序列相似性,尽管它们可能具有相似的结构和生物学特性。序列的抗原性可能以一种微妙且难以理解的方式编码,无法通过序列比对直接识别。由于缺乏与已知来源抗原的相似性,真正新型抗原的发现将受到阻碍。为了克服依赖比对方法的局限性,我们提出了一种新的无比对抗原预测方法,该方法基于将蛋白质序列自动交叉协方差(ACC)转换为主要氨基酸特性的统一向量。
使用细菌、病毒和肿瘤蛋白质数据集来推导预测全蛋白抗原性的模型。每组由100种已知抗原和100种非抗原组成。通过内部留一法交叉验证和使用测试集的外部验证对推导的模型进行测试。另外为每类抗原使用五个训练集来测试抗原与非抗原之间区分的稳定性。这些模型在两种验证中均表现良好,预测准确率为70%至89%。这些模型在一个我们称为VaxiJen的服务器中实现。
VaxiJen是第一个用于独立于序列比对预测保护性抗原的服务器。它的开发是为了仅基于蛋白质的物理化学性质进行抗原分类,而无需借助序列比对。该服务器可以单独使用,也可以与基于比对的预测方法结合使用。它可在以下网址免费在线获取:http://www.jenner.ac.uk/VaxiJen。