Watson James D, Sanderson Steve, Ezersky Alexandra, Savchenko Alexei, Edwards Aled, Orengo Christine, Joachimiak Andrzej, Laskowski Roman A, Thornton Janet M
EMBL--European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK.
J Mol Biol. 2007 Apr 13;367(5):1511-22. doi: 10.1016/j.jmb.2007.01.063. Epub 2007 Jan 30.
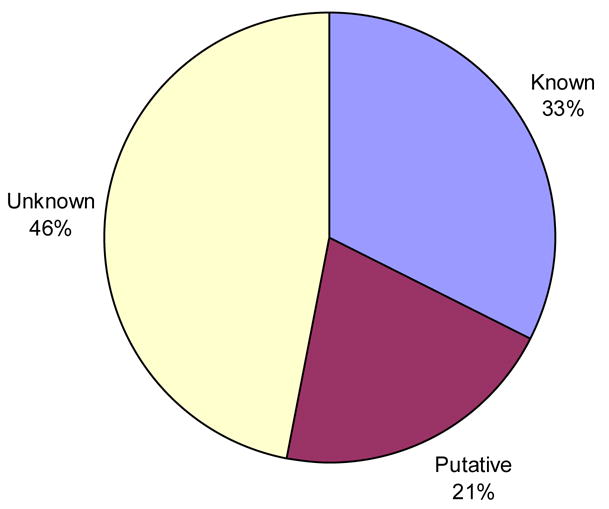
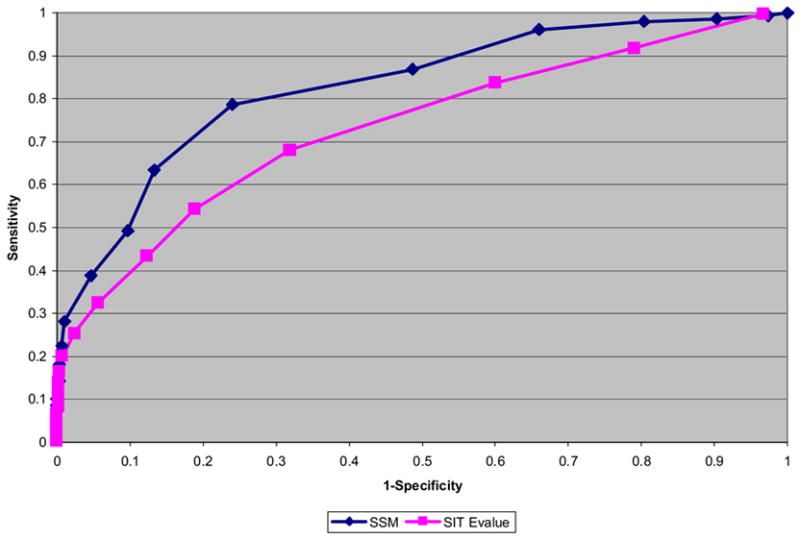
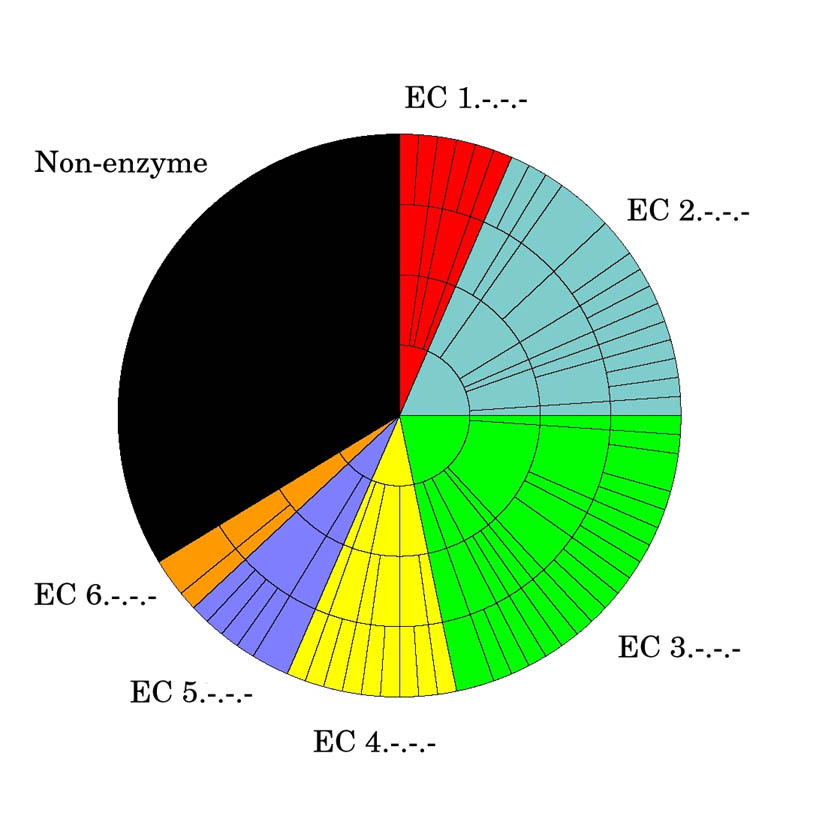
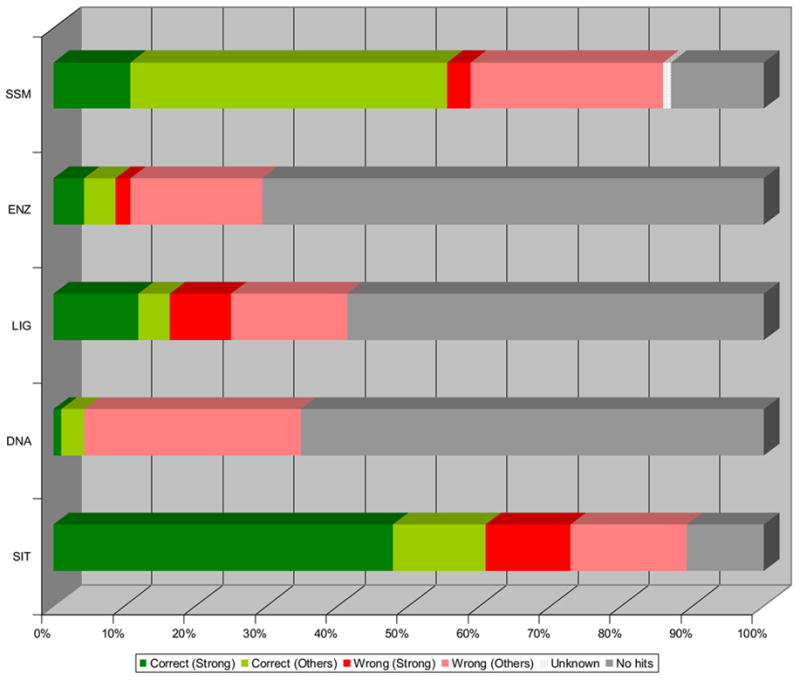
As the global Structural Genomics projects have picked up pace, the number of structures annotated in the Protein Data Bank as hypothetical protein or unknown function has grown significantly. A major challenge now involves the development of computational methods to assign functions to these proteins accurately and automatically. As part of the Midwest Center for Structural Genomics (MCSG) we have developed a fully automated functional analysis server, ProFunc, which performs a battery of analyses on a submitted structure. The analyses combine a number of sequence-based and structure-based methods to identify functional clues. After the first stage of the Protein Structure Initiative (PSI), we review the success of the pipeline and the importance of structure-based function prediction. As a dataset, we have chosen all structures solved by the MCSG during the 5 years of the first PSI. Our analysis suggests that two of the structure-based methods are particularly successful and provide examples of local similarity that is difficult to identify using current sequence-based methods. No one method is successful in all cases, so, through the use of a number of complementary sequence and structural approaches, the ProFunc server increases the chances that at least one method will find a significant hit that can help elucidate function. Manual assessment of the results is a time-consuming process and subject to individual interpretation and human error. We present a method based on the Gene Ontology (GO) schema using GO-slims that can allow the automated assessment of hits with a success rate approaching that of expert manual assessment.
随着全球结构基因组学项目的加速推进,蛋白质数据库中注释为假设蛋白或功能未知的结构数量显著增加。当前的一项重大挑战是开发计算方法,以准确、自动地为这些蛋白质赋予功能。作为中西部结构基因组学中心(MCSG)的一部分,我们开发了一个全自动功能分析服务器ProFunc,它对提交的结构进行一系列分析。这些分析结合了多种基于序列和基于结构的方法来识别功能线索。在蛋白质结构计划(PSI)的第一阶段之后,我们回顾了该流程的成功之处以及基于结构的功能预测的重要性。作为一个数据集,我们选择了MCSG在PSI第一阶段的5年中解析的所有结构。我们的分析表明,两种基于结构的方法特别成功,并提供了使用当前基于序列的方法难以识别的局部相似性示例。没有一种方法在所有情况下都能成功,因此,通过使用多种互补的序列和结构方法,ProFunc服务器增加了至少一种方法找到有助于阐明功能的显著匹配的机会。对结果进行人工评估是一个耗时的过程,并且容易受到个人解读和人为错误的影响。我们提出了一种基于基因本体(GO)模式并使用GO精简版的方法,该方法可以对匹配结果进行自动评估,成功率接近专家人工评估。