Wallace Iain M, Higgins Desmond G
The Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin, Ireland.
BMC Bioinformatics. 2007 Apr 23;8:135. doi: 10.1186/1471-2105-8-135.
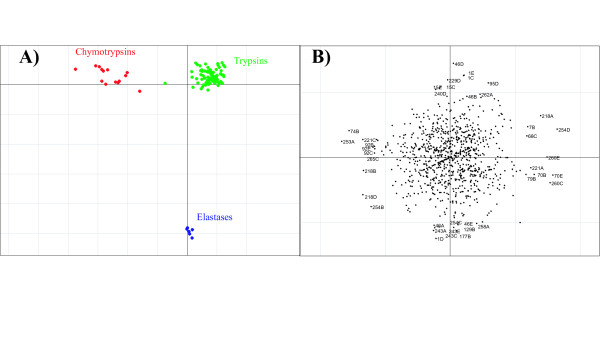
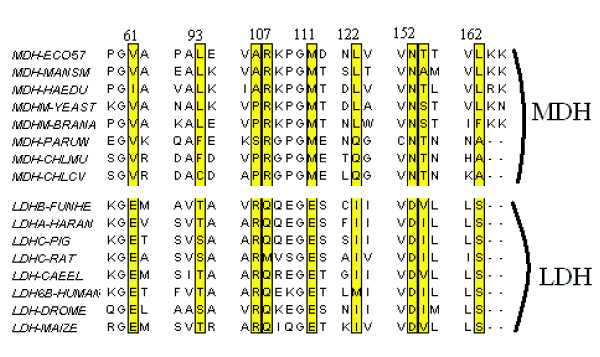
Proteins that evolve from a common ancestor can change functionality over time, and it is important to be able identify residues that cause this change. In this paper we show how a supervised multivariate statistical method, Between Group Analysis (BGA), can be used to identify these residues from families of proteins with different substrate specifities using multiple sequence alignments.
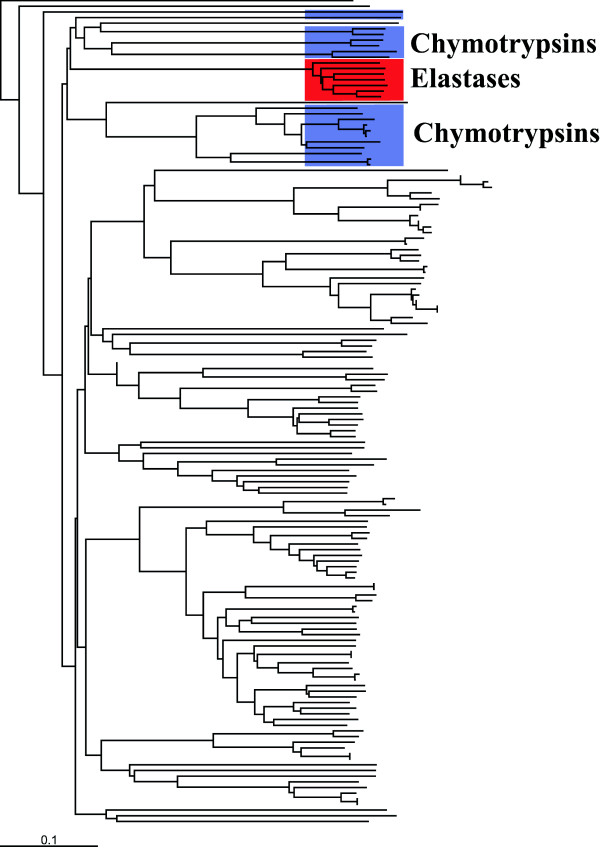
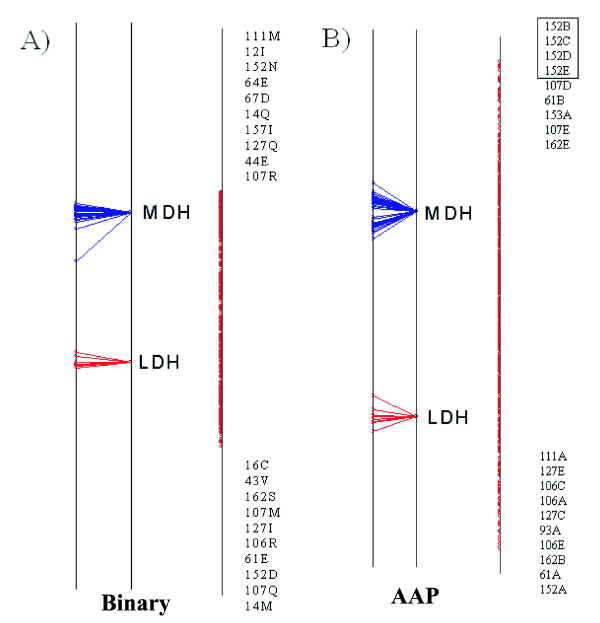
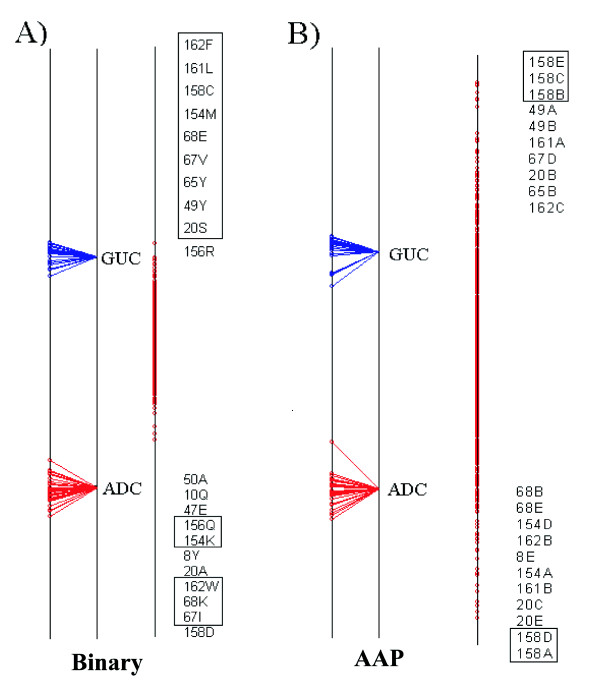
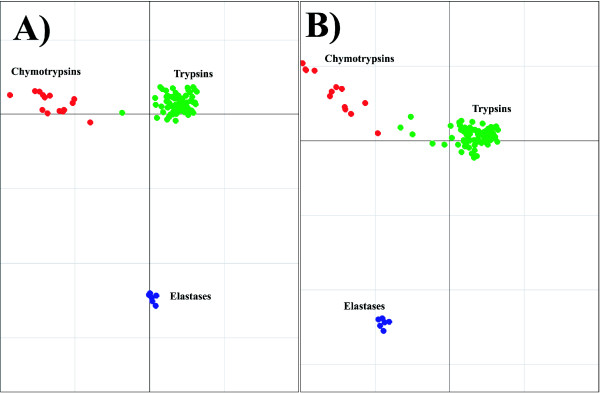
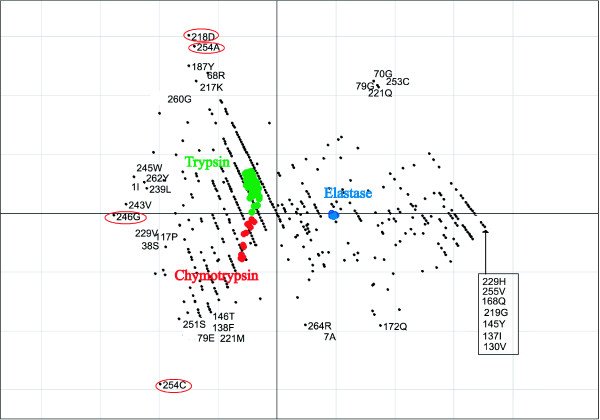
We demonstrate the usefulness of this method on three different test cases. Two of these test cases, the Lactate/Malate dehydrogenase family and Nucleotidyl Cyclases, consist of two functional groups. The other family, Serine Proteases consists of three groups. BGA was used to analyse and visualise these three families using two different encoding schemes for the amino acids.
This overall combination of methods in this paper is powerful and flexible while being computationally very fast and simple. BGA is especially useful because it can be used to analyse any number of functional classes. In the examples we used in this paper, we have only used 2 or 3 classes for demonstration purposes but any number can be used and visualised.
从共同祖先演化而来的蛋白质会随着时间改变功能,识别导致这种变化的残基很重要。在本文中,我们展示了一种监督多元统计方法——组间分析(BGA),如何用于通过多序列比对从具有不同底物特异性的蛋白质家族中识别这些残基。
我们在三个不同的测试案例中证明了该方法的有效性。其中两个测试案例,乳酸/苹果酸脱氢酶家族和核苷酸环化酶,由两个功能组组成。另一个家族,丝氨酸蛋白酶,由三个组组成。使用两种不同的氨基酸编码方案,BGA用于分析和可视化这三个家族。
本文中这些方法的整体组合强大且灵活,同时计算速度非常快且简单。BGA特别有用,因为它可用于分析任意数量的功能类别。在本文使用的示例中,我们仅使用2或3个类别用于演示目的,但可以使用和可视化任意数量的类别。