Stone Eric A, Sidow Arend
Bioinformatics Research Center, North Carolina State University, Raleigh, NC 27695-7566, USA.
BMC Bioinformatics. 2007 Jun 26;8:222. doi: 10.1186/1471-2105-8-222.
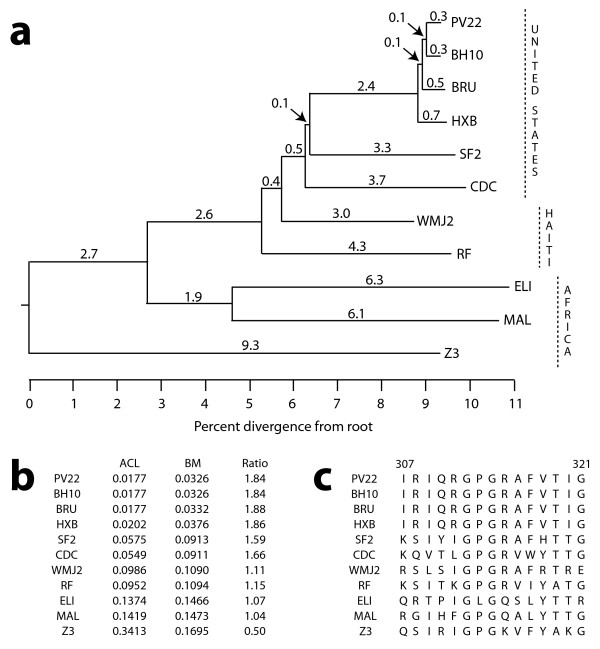
As a consequence of the evolutionary process, data collected from related species tend to be similar. This similarity by descent can obscure subtler signals in the data such as the evidence of constraint on variation due to shared selective pressures. In comparative sequence analysis, for example, sequence similarity is often used to illuminate important regions of the genome, but if the comparison is between closely related species, then similarity is the rule rather than the interesting exception. Furthermore, and perhaps worse yet, the contribution of a divergent third species may be masked by the strong similarity between the other two. Here we propose a remedy that weighs the contribution of each species according to its phylogenetic placement.
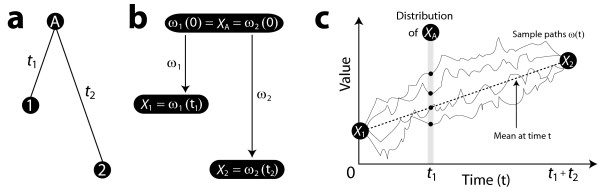
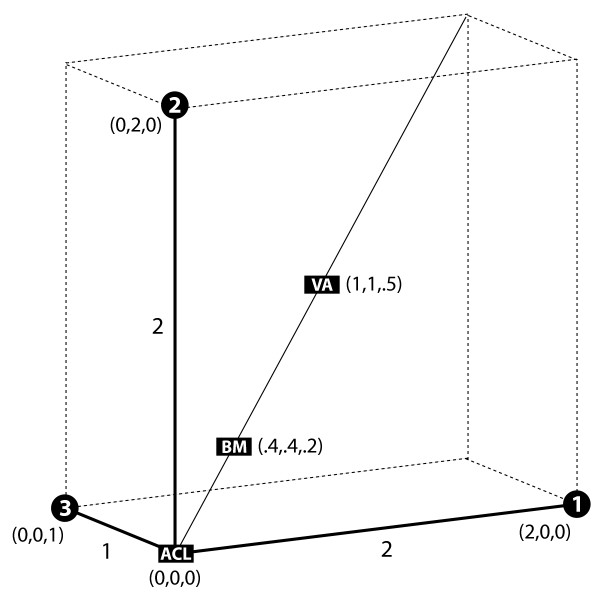
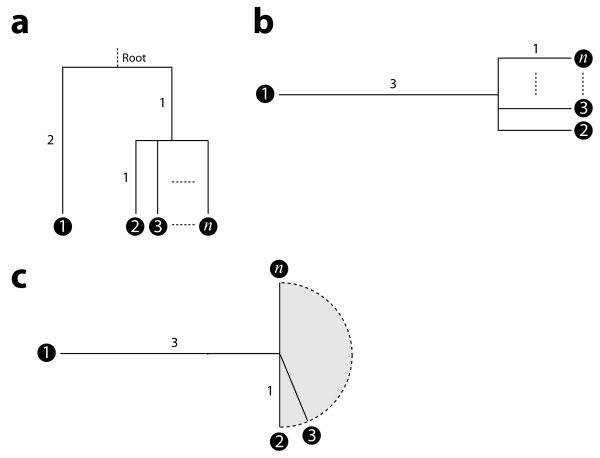
We first solve the problem of summarizing data related by phylogeny, and we explain why an average should operate on the entire evolutionary trajectory that relates the data. This perspective leads to a new approach in which we define the average in terms of the phylogeny, using the data and a stochastic model to obtain a probability on evolutionary trajectories. With the assumption that the data evolve according to a Brownian motion process on the tree, we show that our evolutionary average can be computed as convex combination of the species data. Thus, our approach, called the BranchManager, defines both an average and a novel taxon weighting scheme. We compare the BranchManager to two other methods, demonstrating why it exhibits desirable properties. In doing so, we devise a framework for comparison and introduce the concept of a representative point at which the average is situated.
The BranchManager uses as its representative point the phylogenetic center of mass, a choice which has both intuitive and practical appeal. Because our average is intrinsic to both the dataset and to the phylogeny, we expect it and its corresponding weighting scheme to be useful in all sorts of studies where interspecies data need to be combined. Obvious applications include evolutionary studies of morphology, physiology or behaviour, but quantitative measures such as sequence hydrophobicity and gene expression level are amenable to our approach as well. Other areas of potential impact include motif discovery and vaccine design. A Java implementation of the BranchManager is available for download, as is a script written in the statistical language R.
作为进化过程的结果,从相关物种收集的数据往往具有相似性。这种因亲缘关系而产生的相似性可能会掩盖数据中更细微的信号,比如由于共享选择压力而对变异产生限制的证据。例如,在比较序列分析中,序列相似性常被用于揭示基因组的重要区域,但如果比较是在亲缘关系较近的物种之间进行,那么相似性就是常态而非有趣的例外情况。此外,或许更糟糕的是,第三个分歧物种的贡献可能会被另外两个物种之间的强烈相似性所掩盖。在此,我们提出一种补救方法,即根据每个物种的系统发育位置来权衡其贡献。
我们首先解决了总结系统发育相关数据的问题,并解释了为什么平均值应该作用于与数据相关的整个进化轨迹。这种观点引出了一种新方法,在该方法中,我们根据系统发育来定义平均值,利用数据和一个随机模型来获得进化轨迹上的概率。假设数据在树上按照布朗运动过程进化,我们表明我们的进化平均值可以作为物种数据的凸组合来计算。因此,我们的方法,即分支管理器(BranchManager),既定义了一个平均值,也定义了一种新颖的分类单元加权方案。我们将分支管理器与其他两种方法进行比较,展示了它为何具有理想的特性。在此过程中,我们设计了一个比较框架,并引入了平均值所在的代表点的概念。
分支管理器以系统发育质心作为其代表点,这一选择兼具直观性和实用性。由于我们的平均值对于数据集和系统发育都是内在的,我们期望它及其相应的加权方案在所有需要组合种间数据的各类研究中都有用。明显的应用包括形态学、生理学或行为的进化研究,但诸如序列疏水性和基因表达水平等定量测量也适用于我们的方法。其他潜在影响领域包括基序发现和疫苗设计。分支管理器的Java实现版本可供下载,还有一个用统计语言R编写的脚本也可供下载。