Kmiecik Sebastian, Gront Dominik, Kolinski Andrzej
University of Warsaw, Faculty of Chemistry, Pasteura 1, Warsaw, Poland.
BMC Struct Biol. 2007 Jun 29;7:43. doi: 10.1186/1472-6807-7-43.
Although experimental methods for determining protein structure are providing high resolution structures, they cannot keep the pace at which amino acid sequences are resolved on the scale of entire genomes. For a considerable fraction of proteins whose structures will not be determined experimentally, computational methods can provide valuable information. The value of structural models in biological research depends critically on their quality. Development of high-accuracy computational methods that reliably generate near-experimental quality structural models is an important, unsolved problem in the protein structure modeling.
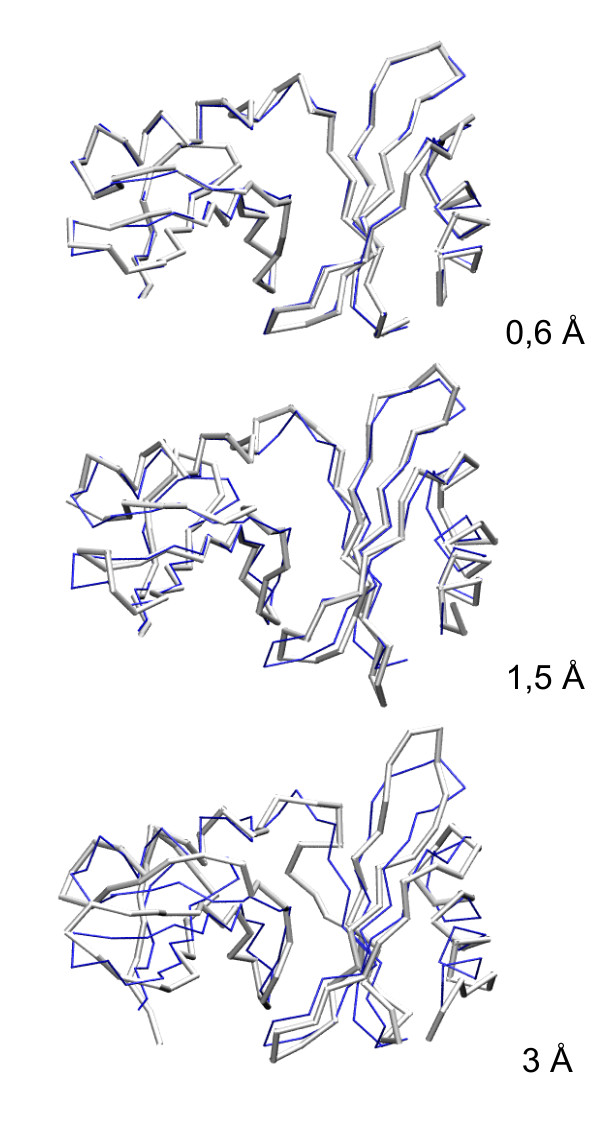
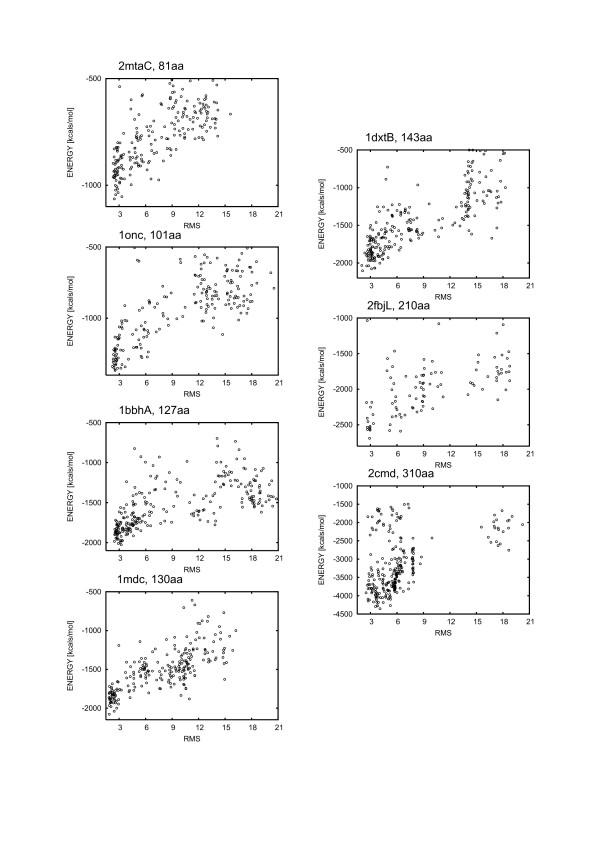
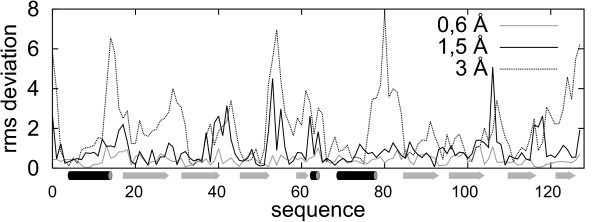
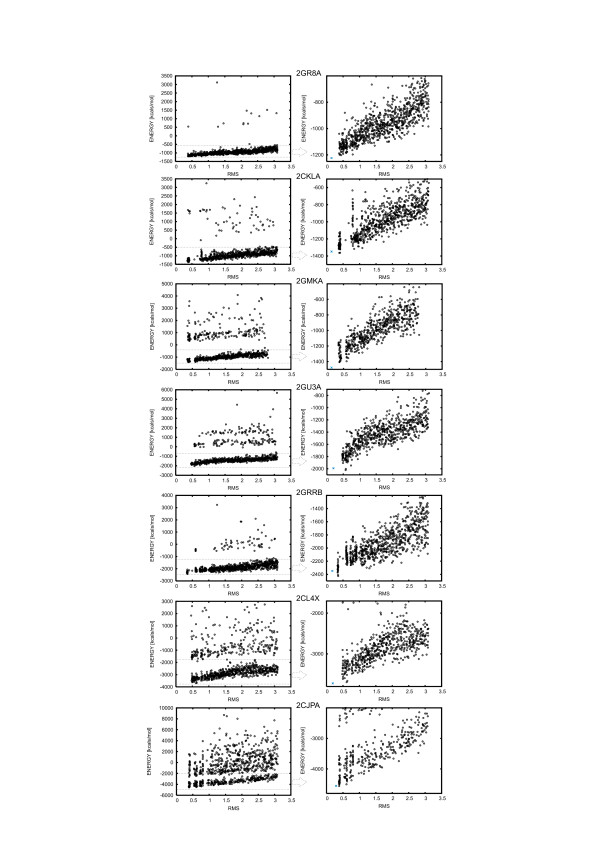
Large sets of structural decoys have been generated using reduced conformational space protein modeling tool CABS. Subsequently, the reduced models were subject to all-atom reconstruction. Then, the resulting detailed models were energy-minimized using state-of-the-art all-atom force field, assuming fixed positions of the alpha carbons. It has been shown that a very short minimization leads to the proper ranking of the quality of the models (distance from the native structure), when the all-atom energy is used as the ranking criterion. Additionally, we performed test on medium and low accuracy decoys built via classical methods of comparative modeling. The test placed our model evaluation procedure among the state-of-the-art protein model assessment methods.
These test computations show that a large scale high resolution protein structure prediction is possible, not only for small but also for large protein domains, and that it should be based on a hierarchical approach to the modeling protocol. We employed Molecular Mechanics with fixed alpha carbons to rank-order the all-atom models built on the scaffolds of the reduced models. Our tests show that a physic-based approach, usually considered computationally too demanding for large-scale applications, can be effectively used in such studies.
尽管用于确定蛋白质结构的实验方法能够提供高分辨率结构,但在解析整个基因组规模的氨基酸序列方面,它们已无法跟上步伐。对于相当一部分其结构无法通过实验确定的蛋白质而言,计算方法能够提供有价值的信息。生物研究中结构模型的价值关键取决于其质量。开发能够可靠生成接近实验质量结构模型的高精度计算方法,是蛋白质结构建模中一个重要的、尚未解决的问题。
使用简化构象空间蛋白质建模工具CABS生成了大量的结构诱饵。随后,对简化模型进行全原子重建。然后,使用最先进的全原子力场对得到的详细模型进行能量最小化,假设α碳原子位置固定。结果表明,当将全原子能量用作排序标准时,非常短时间的最小化就能使模型质量(与天然结构的距离)得到正确排序。此外,我们对通过经典比较建模方法构建的中等精度和低精度诱饵进行了测试。该测试将我们的模型评估程序置于最先进的蛋白质模型评估方法之列。
这些测试计算表明,不仅对于小的蛋白质结构域,而且对于大的蛋白质结构域,大规模高分辨率蛋白质结构预测都是可能的,并且应该基于对建模协议的分层方法。我们采用固定α碳原子的分子力学方法对基于简化模型支架构建的全原子模型进行排序。我们的测试表明,一种通常被认为对大规模应用计算要求过高的基于物理的方法,可以有效地用于此类研究。