Teschendorff Andrew E, Journée Michel, Absil Pierre A, Sepulchre Rodolphe, Caldas Carlos
Breast Cancer Functional Genomics Laboratory, Cancer Research UK Cambridge Research Institute, Cambridge, United Kingdom.
PLoS Comput Biol. 2007 Aug;3(8):e161. doi: 10.1371/journal.pcbi.0030161. Epub 2007 Jun 29.
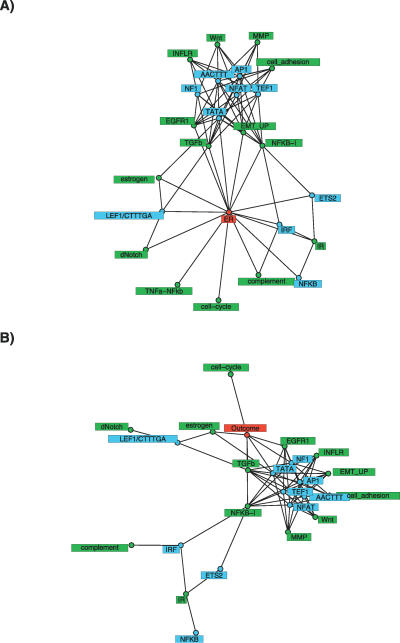
The quantity of mRNA transcripts in a cell is determined by a complex interplay of cooperative and counteracting biological processes. Independent Component Analysis (ICA) is one of a few number of unsupervised algorithms that have been applied to microarray gene expression data in an attempt to understand phenotype differences in terms of changes in the activation/inhibition patterns of biological pathways. While the ICA model has been shown to outperform other linear representations of the data such as Principal Components Analysis (PCA), a validation using explicit pathway and regulatory element information has not yet been performed. We apply a range of popular ICA algorithms to six of the largest microarray cancer datasets and use pathway-knowledge and regulatory-element databases for validation. We show that ICA outperforms PCA and clustering-based methods in that ICA components map closer to known cancer-related pathways, regulatory modules, and cancer phenotypes. Furthermore, we identify cancer signalling and oncogenic pathways and regulatory modules that play a prominent role in breast cancer and relate the differential activation patterns of these to breast cancer phenotypes. Importantly, we find novel associations linking immune response and epithelial-mesenchymal transition pathways with estrogen receptor status and histological grade, respectively. In addition, we find associations linking the activity levels of biological pathways and transcription factors (NF1 and NFAT) with clinical outcome in breast cancer. ICA provides a framework for a more biologically relevant interpretation of genomewide transcriptomic data. Adopting ICA as the analysis tool of choice will help understand the phenotype-pathway relationship and thus help elucidate the molecular taxonomy of heterogeneous cancers and of other complex genetic diseases.
细胞中mRNA转录本的数量由协同和拮抗生物学过程的复杂相互作用决定。独立成分分析(ICA)是少数几种无监督算法之一,已应用于微阵列基因表达数据,试图根据生物途径激活/抑制模式的变化来理解表型差异。虽然ICA模型已被证明优于数据的其他线性表示方法,如主成分分析(PCA),但尚未使用明确的途径和调控元件信息进行验证。我们将一系列流行的ICA算法应用于六个最大的微阵列癌症数据集,并使用途径知识和调控元件数据库进行验证。我们表明,ICA在性能上优于PCA和基于聚类的方法,因为ICA成分更接近已知的癌症相关途径、调控模块和癌症表型。此外,我们确定了在乳腺癌中起重要作用的癌症信号传导和致癌途径以及调控模块,并将这些途径的差异激活模式与乳腺癌表型联系起来。重要的是,我们发现了新的关联,分别将免疫反应和上皮-间质转化途径与雌激素受体状态和组织学分级联系起来。此外,我们发现生物途径和转录因子(NF1和NFAT)的活性水平与乳腺癌临床结果之间存在关联。ICA为全基因组转录组数据提供了一个更具生物学相关性的解释框架。采用ICA作为首选分析工具将有助于理解表型-途径关系,从而有助于阐明异质性癌症和其他复杂遗传疾病的分子分类。