University of California San Diego, San Diego, USA.
BMC Bioinformatics. 2021 Dec 8;22(1):584. doi: 10.1186/s12859-021-04497-7.
Independent component analysis is an unsupervised machine learning algorithm that separates a set of mixed signals into a set of statistically independent source signals. Applied to high-quality gene expression datasets, independent component analysis effectively reveals both the source signals of the transcriptome as co-regulated gene sets, and the activity levels of the underlying regulators across diverse experimental conditions. Two major variables that affect the final gene sets are the diversity of the expression profiles contained in the underlying data, and the user-defined number of independent components, or dimensionality, to compute. Availability of high-quality transcriptomic datasets has grown exponentially as high-throughput technologies have advanced; however, optimal dimensionality selection remains an open question.
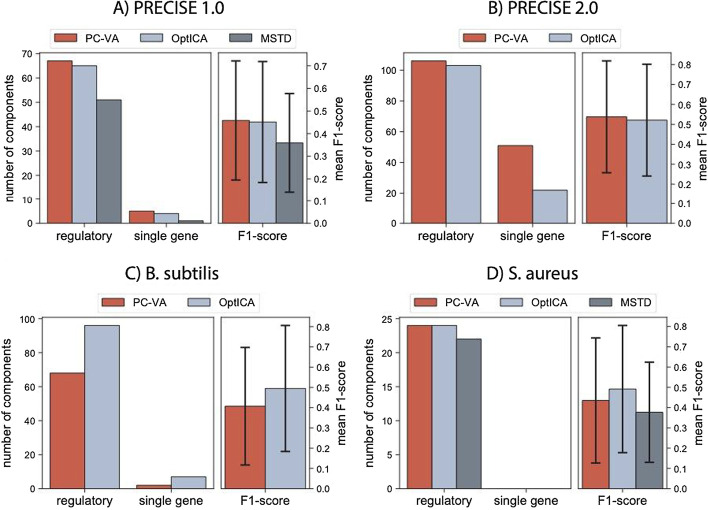
We computed independent components across a range of dimensionalities for four gene expression datasets with varying dimensions (both in terms of number of genes and number of samples). We computed the correlation between independent components across different dimensionalities to understand how the overall structure evolves as the number of user-defined components increases. We then measured how well the resulting gene clusters reflected known regulatory mechanisms, and developed a set of metrics to assess the accuracy of the decomposition at a given dimension.
We found that over-decomposition results in many independent components dominated by a single gene, whereas under-decomposition results in independent components that poorly capture the known regulatory structure. From these results, we developed a new method, called OptICA, for finding the optimal dimensionality that controls for both over- and under-decomposition. Specifically, OptICA selects the highest dimension that produces a low number of components that are dominated by a single gene. We show that OptICA outperforms two previously proposed methods for selecting the number of independent components across four transcriptomic databases of varying sizes.
OptICA avoids both over-decomposition and under-decomposition of transcriptomic datasets resulting in the best representation of the organism's underlying transcriptional regulatory network.
独立成分分析是一种无监督机器学习算法,可将一组混合信号分离成一组统计上独立的源信号。将其应用于高质量基因表达数据集,可有效揭示转录组的源信号作为共同调节基因集,以及在不同实验条件下潜在调节因子的活性水平。影响最终基因集的两个主要变量是基础数据中包含的表达谱的多样性,以及要计算的独立成分(或维度)的用户定义数量。随着高通量技术的进步,高质量转录组数据集的可用性呈指数级增长;然而,最佳维度选择仍然是一个悬而未决的问题。
我们针对四个具有不同维度(基因数量和样本数量)的基因表达数据集,在一系列维度上计算独立成分。我们计算了不同维度之间的独立成分之间的相关性,以了解随着用户定义组件数量的增加,整体结构如何演变。然后,我们测量了由此产生的基因簇如何反映已知的调节机制,并开发了一组指标来评估在给定维度下分解的准确性。
我们发现过度分解会导致许多由单个基因主导的独立成分,而欠分解则会导致独立成分无法很好地捕捉已知的调节结构。根据这些结果,我们开发了一种新的方法,称为 OptICA,用于找到控制过度和欠分解的最佳维度。具体来说,OptICA 选择产生数量较少的组件的最高维度,这些组件由单个基因主导。我们表明,OptICA 在四个不同大小的转录组数据库中选择独立成分数量的两种先前提出的方法表现更好。
OptICA 避免了转录组数据集的过度分解和欠分解,从而最佳地表示了生物体潜在的转录调节网络。