Swart Marcel, van der Wijst Tushar, Fonseca Guerra Célia, Bickelhaupt F Matthias
Theoretische Chemie, Vrije Universiteit, De Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands.
J Mol Model. 2007 Dec;13(12):1245-57. doi: 10.1007/s00894-007-0239-y. Epub 2007 Sep 15.
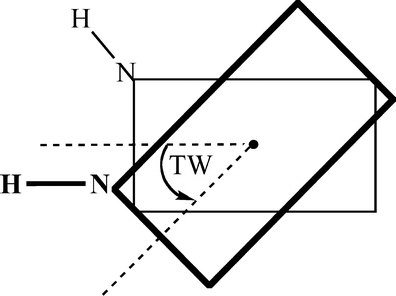
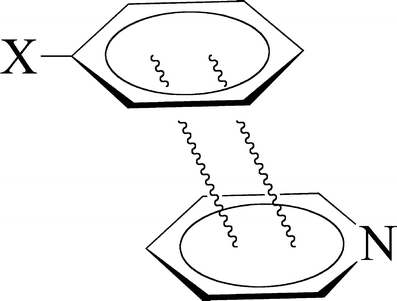
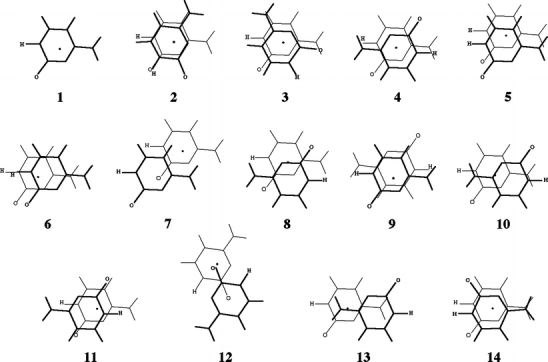
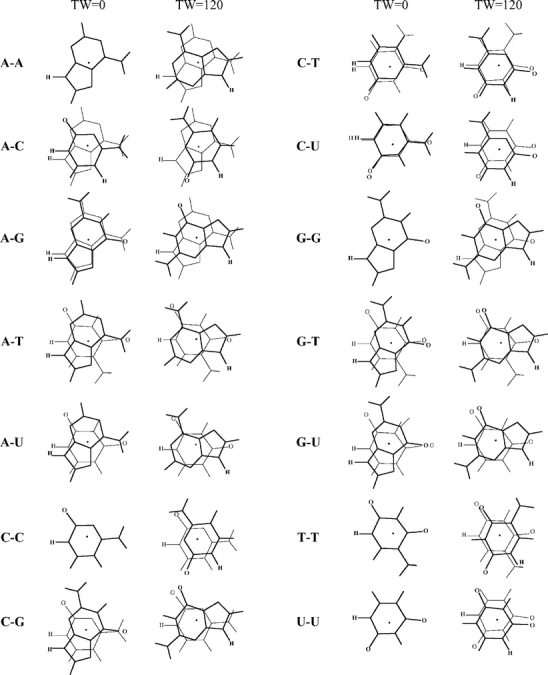
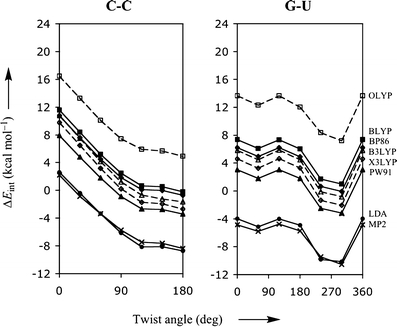
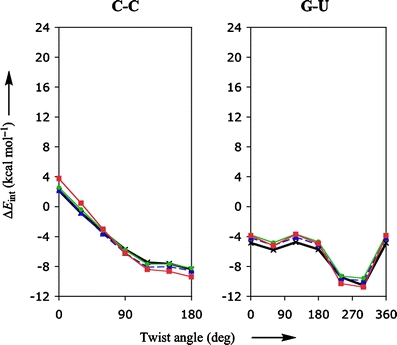
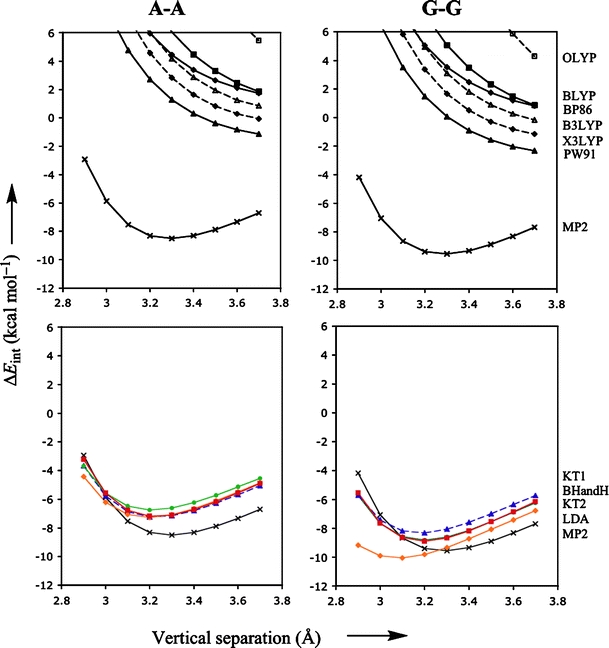
Through comparison with ab initio reference data, we have evaluated the performance of various density functionals for describing pi-pi interactions as a function of the geometry between two stacked benzenes or benzene analogs, between two stacked DNA bases, and between two stacked Watson-Crick pairs. Our main purpose is to find a robust and computationally efficient density functional to be used specifically and only for describing pi-pi stacking interactions in DNA and other biological molecules in the framework of our recently developed QM/QM approach "QUILD". In line with previous studies, most standard density functionals recover, at best, only part of the favorable stacking interactions. An exception is the new KT1 functional, which correctly yields bound pi-stacked structures. Surprisingly, a similarly good performance is achieved with the computationally very robust and efficient local density approximation (LDA). Furthermore, we show that classical electrostatic interactions determine the shape and depth of the pi-pi stacking potential energy surface.
通过与从头算参考数据进行比较,我们评估了各种密度泛函在描述π-π相互作用时的性能,这些相互作用是两个堆叠苯或苯类似物之间、两个堆叠DNA碱基之间以及两个堆叠的沃森-克里克碱基对之间几何结构的函数。我们的主要目的是找到一种稳健且计算效率高的密度泛函,专门用于在我们最近开发的QM/QM方法“QUILD”框架下描述DNA和其他生物分子中的π-π堆积相互作用。与之前的研究一致,大多数标准密度泛函最多只能恢复部分有利的堆积相互作用。一个例外是新的KT1泛函,它能正确地产生结合的π堆积结构。令人惊讶的是,计算上非常稳健且高效的局域密度近似(LDA)也能实现类似的良好性能。此外,我们表明经典静电相互作用决定了π-π堆积势能面的形状和深度。