Morton Richard A, Morton Brian R
Department of Biology, McMaster University, 1280 Main Street West, Hamilton ON L8S 4K1, Canada.
BMC Genomics. 2007 Oct 12;8:369. doi: 10.1186/1471-2164-8-369.
Many bacterial chromosomes display nucleotide asymmetry, or skew, between the leading and lagging strands of replication. Mutational differences between these strands result in an overall pattern of skew that is centered about the origin of replication. Such a pattern could also arise from selection coupled with a bias for genes coded on the leading strand. The relative contributions of selection and mutation in producing compositional skew are largely unknown.
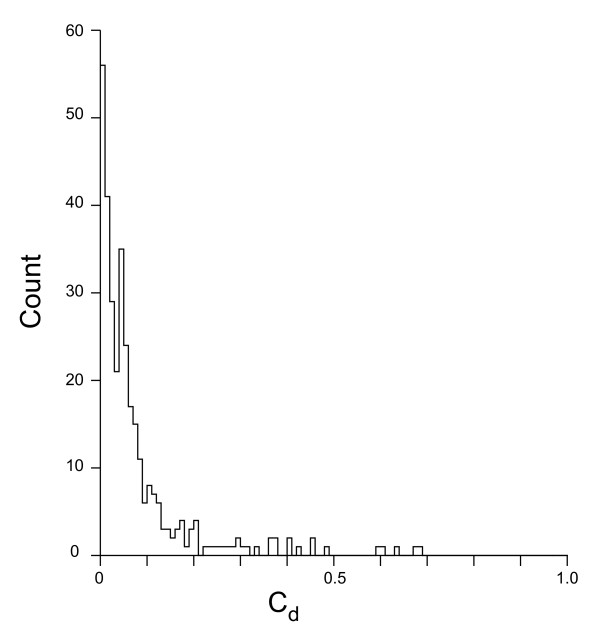
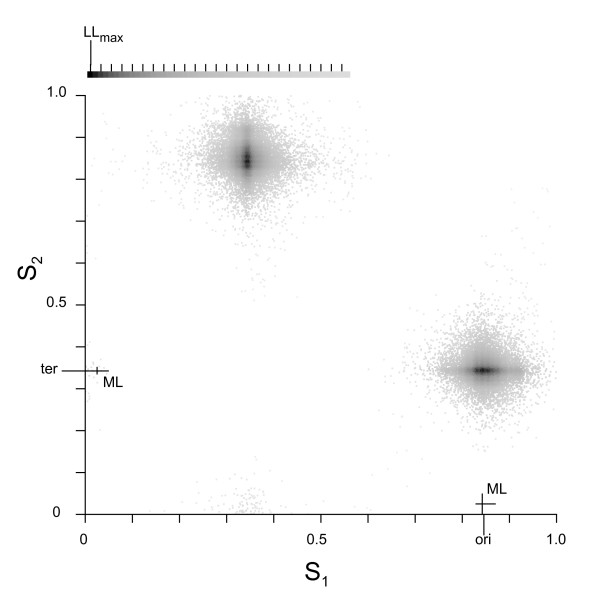
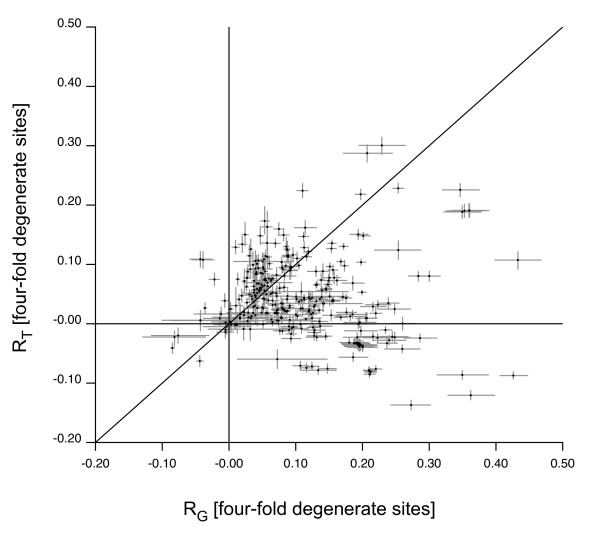
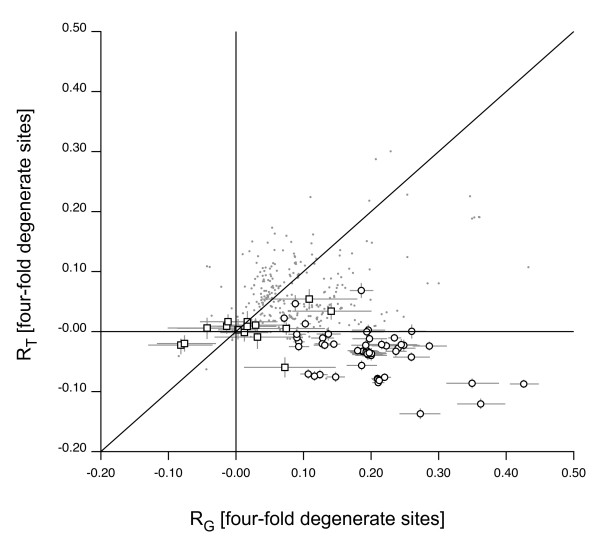
We describe a model to quantify the contribution of mutational differences between the leading and lagging strands in producing replication-induced skew. When the origin and terminus of replication are known, the model can be used to estimate the relative accumulation of G over C and of A over T on the leading strand due to replication effects in a chromosome with bidirectional replication arms. The model may also be implemented in a maximum likelihood framework to estimate the locations of origin and terminus. We find that our estimations for the origin and terminus agree very well with the location of genes that are thought to be associated with the replication origin. This indicates that our model provides an accurate, objective method of determining the replication arms and also provides support for the hypothesis that these genes represent an ancestral cluster of origin-associated genes.
The model has several advantages over other methods of analyzing genome skew. First, it quantifies the role of mutation in generating skew so that its effect on composition, for example codon bias, can be assessed. Second, it provides an objective method for locating origin and terminus, one that is based on chromosome-wide accumulation of leading vs lagging strand nucleotide differences. Finally, the model has the potential to be utilized in a maximum likelihood framework in order to analyze the effect of chromosome rearrangements on nucleotide composition.
许多细菌染色体在复制的前导链和后随链之间表现出核苷酸不对称性,即偏斜。这些链之间的突变差异导致了以复制起点为中心的整体偏斜模式。这种模式也可能源于选择以及对在前导链上编码的基因的偏好。在产生组成性偏斜方面,选择和突变的相对贡献在很大程度上尚不清楚。
我们描述了一个模型,用于量化前导链和后随链之间的突变差异在产生复制诱导偏斜中的作用。当复制起点和终点已知时,该模型可用于估计在具有双向复制臂的染色体中,由于复制效应,前导链上G相对于C以及A相对于T的相对积累。该模型也可以在最大似然框架中实现,以估计起点和终点的位置。我们发现,我们对起点和终点的估计与被认为与复制起点相关的基因的位置非常吻合。这表明我们的模型提供了一种准确、客观的方法来确定复制臂,也为这些基因代表起源相关基因的祖先簇这一假设提供了支持。
该模型相对于其他分析基因组偏斜的方法具有几个优点。首先,它量化了突变在产生偏斜中的作用,从而可以评估其对组成的影响,例如密码子偏好。其次,它提供了一种客观的方法来定位起点和终点,该方法基于全染色体范围内前导链与后随链核苷酸差异的积累。最后,该模型有潜力在最大似然框架中用于分析染色体重排对核苷酸组成的影响。