Cheng Jianlin
Department of Computer Science, Informatics Institute, University of Missouri, Columbia, MO 65211-2060, USA.
BMC Struct Biol. 2008 Mar 17;8:18. doi: 10.1186/1472-6807-8-18.
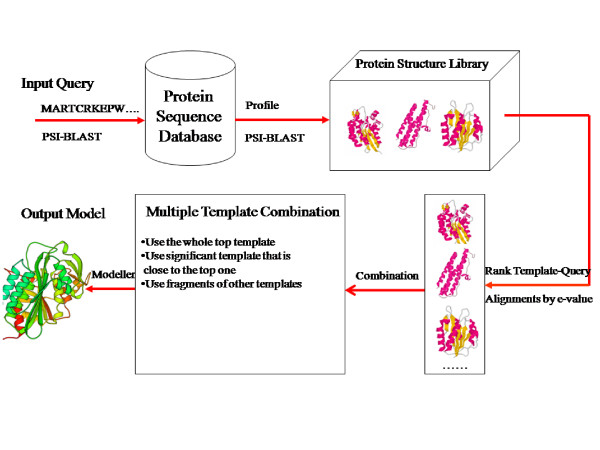
Multiple protein templates are commonly used in manual protein structure prediction. However, few automated algorithms of selecting and combining multiple templates are available.
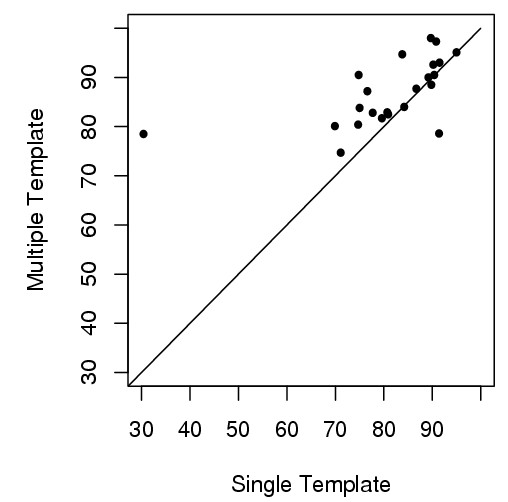
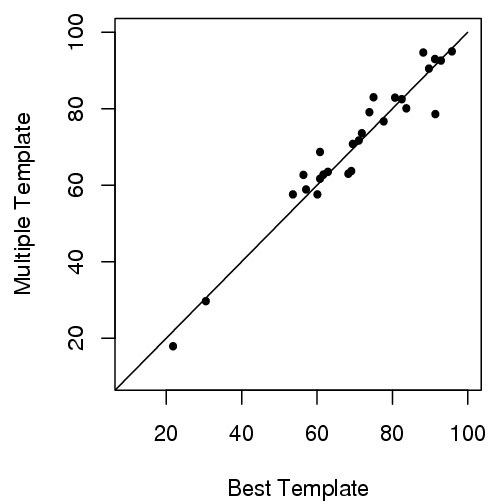
Here we develop an effective multi-template combination algorithm for protein comparative modeling. The algorithm selects templates according to the similarity significance of the alignments between template and target proteins. It combines the whole template-target alignments whose similarity significance score is close to that of the top template-target alignment within a threshold, whereas it only takes alignment fragments from a less similar template-target alignment that align with a sizable uncovered region of the target. We compare the algorithm with the traditional method of using a single top template on the 45 comparative modeling targets (i.e. easy template-based modeling targets) used in the seventh edition of Critical Assessment of Techniques for Protein Structure Prediction (CASP7). The multi-template combination algorithm improves the GDT-TS scores of predicted models by 6.8% on average. The statistical analysis shows that the improvement is significant (p-value < 10-4). Compared with the ideal approach that always uses the best template, the multi-template approach yields only slightly better performance. During the CASP7 experiment, the preliminary implementation of the multi-template combination algorithm (FOLDpro) was ranked second among 67 servers in the category of high-accuracy structure prediction in terms of GDT-TS measure.
We have developed a novel multi-template algorithm to improve protein comparative modeling.
在蛋白质结构预测中,多模板法是一种常用的人工预测方法。然而,目前能自动选择并组合多个模板的算法却很少。
我们开发了一种有效的蛋白质比较建模多模板组合算法。该算法根据模板与目标蛋白比对的相似性显著性来选择模板。它会组合那些相似性显著性得分在阈值范围内且接近最优模板-目标比对得分的完整模板-目标比对,而对于相似度较低的模板-目标比对,仅采用与目标蛋白较大未覆盖区域比对的比对片段。我们将该算法与在蛋白质结构预测技术关键评估(CASP7)第七版中使用的45个比较建模目标(即基于模板的简单建模目标)上使用单一最优模板的传统方法进行了比较。多模板组合算法使预测模型的GDT-TS得分平均提高了6.8%。统计分析表明这种提高具有显著性(p值<10-4)。与始终使用最佳模板的理想方法相比,多模板方法的性能仅略优。在CASP7实验中,多模板组合算法的初步实现(FOLDpro)在基于GDT-TS度量的高精度结构预测类别中,在67个服务器中排名第二。
我们开发了一种新型多模板算法来改进蛋白质比较建模。