Mao Chunhong, Evans Clive, Jensen Roderick V, Sobral Bruno Ws
Virginia Bioinformatics Institute, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA.
BMC Microbiol. 2008 May 2;8:72. doi: 10.1186/1471-2180-8-72.
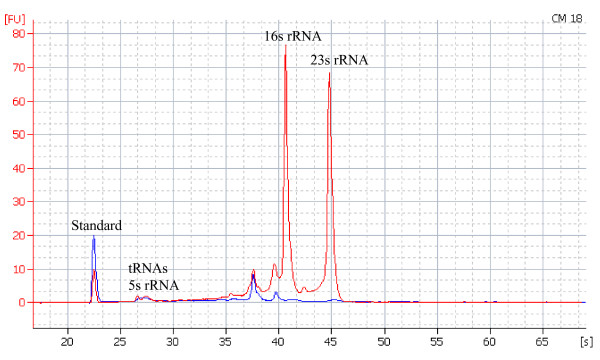
Sinorhizobium meliloti is an agriculturally important model symbiont. There is an ongoing need to update and improve its genome annotation. In this study, we used a high-throughput pyrosequencing approach to sequence the transcriptome of S. meliloti, and search for new bacterial genes missed in the previous genome annotation. This is the first report of sequencing a bacterial transcriptome using the pyrosequencing technology.
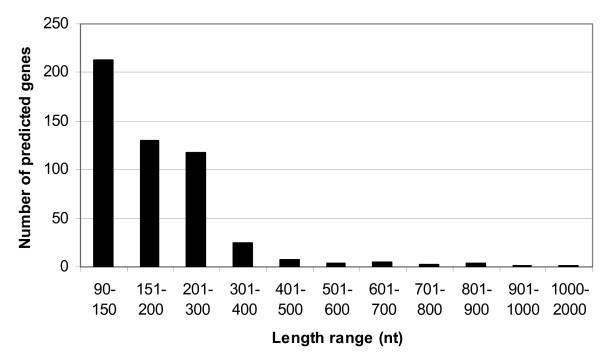
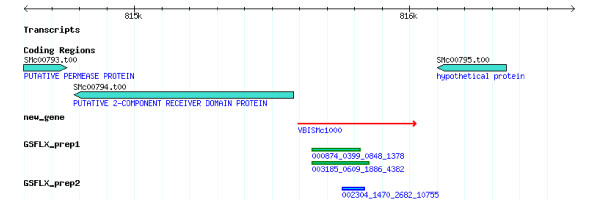
Our pilot sequencing run generated 19,005 reads with an average length of 136 nucleotides per read. From these data, we identified 20 new genes. These new gene transcripts were confirmed by RT-PCR and their possible functions were analyzed.
Our results indicate that high-throughput sequence analysis of bacterial transcriptomes is feasible and next-generation sequencing technologies will greatly facilitate the discovery of new genes and improve genome annotation.
苜蓿中华根瘤菌是一种在农业上具有重要意义的模式共生菌。持续需要更新和改进其基因组注释。在本研究中,我们使用高通量焦磷酸测序方法对苜蓿中华根瘤菌的转录组进行测序,并寻找先前基因组注释中遗漏的新细菌基因。这是使用焦磷酸测序技术对细菌转录组进行测序的首篇报道。
我们的初步测序运行产生了19,005条读数,每条读数的平均长度为136个核苷酸。从这些数据中,我们鉴定出20个新基因。这些新基因转录本通过逆转录聚合酶链反应得到证实,并对其可能的功能进行了分析。
我们的结果表明,细菌转录组的高通量序列分析是可行的,新一代测序技术将极大地促进新基因的发现并改进基因组注释。