Nolte Ilja M, de Vries André R, Spijker Geert T, Jansen Ritsert C, Brinza Dumitru, Zelikovsky Alexander, Te Meerman Gerard J
Department of Epidemiology, University Medical Center Groningen, University of Groningen, Hanzeplein 1, 9713 GZ Groningen, the Netherlands.
Department of Genetics, University Medical Center Groningen, University of Groningen, Hanzeplein 1, 9713 GZ Groningen, the Netherlands.
BMC Proc. 2007;1 Suppl 1(Suppl 1):S129. doi: 10.1186/1753-6561-1-s1-s129. Epub 2007 Dec 18.
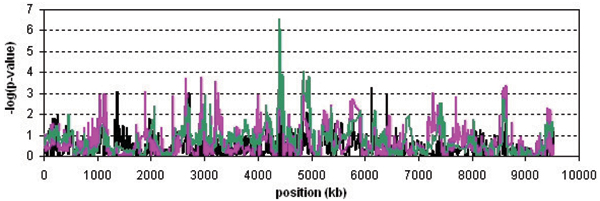
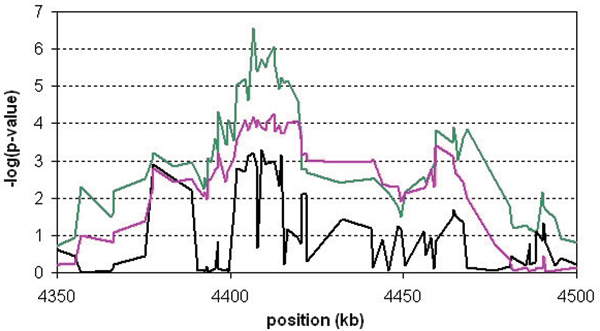
We propose two new haplotype-sharing methods for identifying disease loci: the haplotype sharing statistic (HSS), which compares length of shared haplotypes between cases and controls, and the CROSS test, which tests whether a case and a control haplotype show less sharing than two random haplotypes. The significance of the HSS is determined using a variance estimate from the theory of U-statistics, whereas the significance of the CROSS test is estimated from a sequential randomization procedure. Both methods are fast and hence practical, even for whole-genome screens with high marker densities. We analyzed data sets of Problems 2 and 3 of Genetic Analysis Workshop 15 and compared HSS and CROSS to conventional association methods. Problem 2 provided a data set of 2300 single-nucleotide polymorphisms (SNPs) in a 10-Mb region of chromosome 18q, which had shown linkage evidence for rheumatoid arthritis. The CROSS test detected a significant association at approximately position 4407 kb. This was supported by single-marker association and HSS. The CROSS test outperformed them both with respect to significance level and signal-to-noise ratio. A 20-kb candidate region could be identified. Problem 3 provided a simulated 10 k SNP data set covering the whole genome. Three known candidate regions for rheumatoid arthritis were detected. Again, the CROSS test gave the most significant results. Furthermore, both the HSS and the CROSS showed better fine-mapping accuracy than straightforward haplotype association. In conclusion, haplotype sharing methods, particularly the CROSS test, show great promise for identifying disease gene loci.
单倍型共享统计量(HSS),用于比较病例组和对照组之间共享单倍型的长度;以及CROSS检验,用于检验病例组和对照组的单倍型是否比两个随机单倍型显示出更少的共享。HSS的显著性是使用U统计量理论中的方差估计来确定的,而CROSS检验的显著性是通过顺序随机化程序来估计的。这两种方法都很快,因此即使对于具有高标记密度的全基因组筛查也是实用的。我们分析了遗传分析研讨会15的问题2和问题3的数据集,并将HSS和CROSS与传统关联方法进行了比较。问题2提供了一个位于18号染色体10 Mb区域的2300个单核苷酸多态性(SNP)的数据集,该区域已显示出类风湿性关节炎的连锁证据。CROSS检验在大约4407 kb的位置检测到显著关联。这得到了单标记关联和HSS的支持。在显著性水平和信噪比方面,CROSS检验均优于这两者。可以识别出一个20 kb的候选区域。问题3提供了一个覆盖整个基因组的模拟10 k SNP数据集。检测到类风湿性关节炎的三个已知候选区域。同样,CROSS检验给出了最显著的结果。此外,HSS和CROSS在精细定位准确性方面均优于直接的单倍型关联。总之,单倍型共享方法,特别是CROSS检验,在识别疾病基因座方面显示出巨大的前景。