Hoggart Clive J, Whittaker John C, De Iorio Maria, Balding David J
Department of Epidemiology and Public Health, Imperial College, London, United Kingdom.
PLoS Genet. 2008 Jul 25;4(7):e1000130. doi: 10.1371/journal.pgen.1000130.
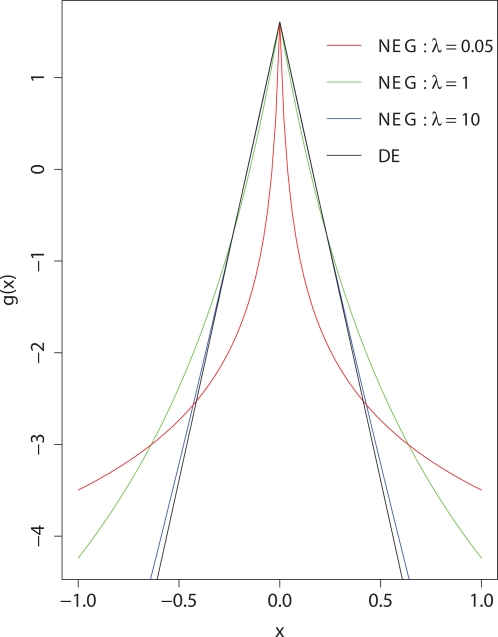
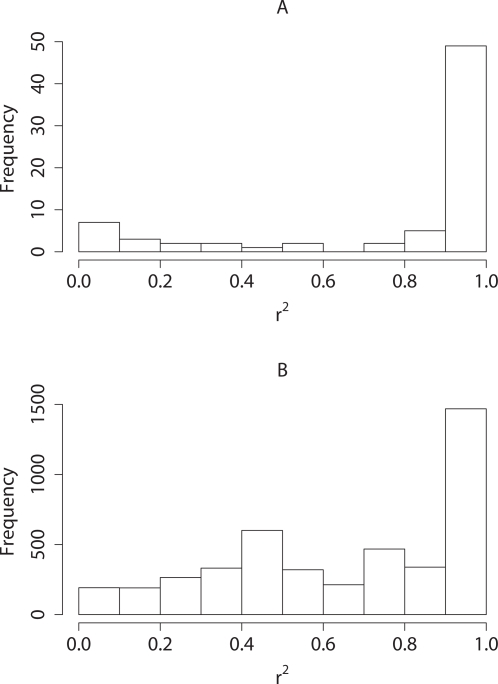
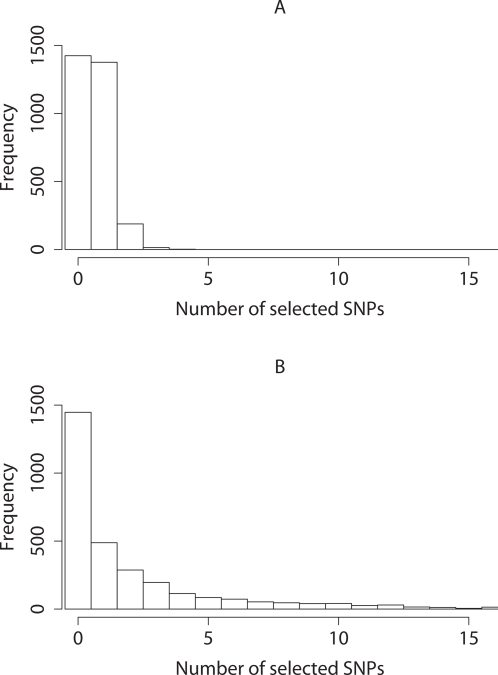
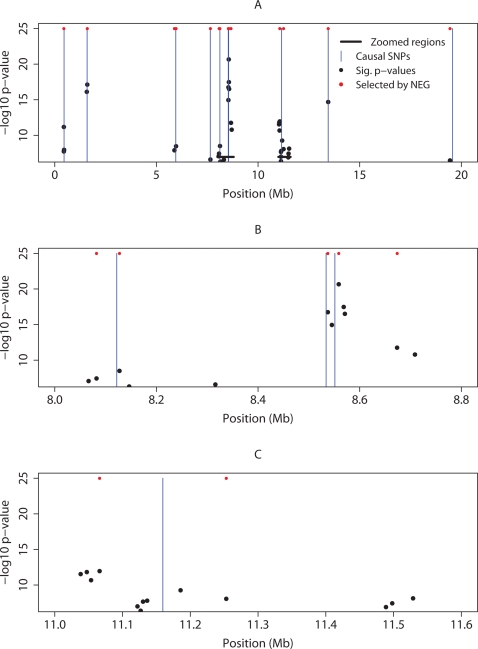
Testing one SNP at a time does not fully realise the potential of genome-wide association studies to identify multiple causal variants, which is a plausible scenario for many complex diseases. We show that simultaneous analysis of the entire set of SNPs from a genome-wide study to identify the subset that best predicts disease outcome is now feasible, thanks to developments in stochastic search methods. We used a Bayesian-inspired penalised maximum likelihood approach in which every SNP can be considered for additive, dominant, and recessive contributions to disease risk. Posterior mode estimates were obtained for regression coefficients that were each assigned a prior with a sharp mode at zero. A non-zero coefficient estimate was interpreted as corresponding to a significant SNP. We investigated two prior distributions and show that the normal-exponential-gamma prior leads to improved SNP selection in comparison with single-SNP tests. We also derived an explicit approximation for type-I error that avoids the need to use permutation procedures. As well as genome-wide analyses, our method is well-suited to fine mapping with very dense SNP sets obtained from re-sequencing and/or imputation. It can accommodate quantitative as well as case-control phenotypes, covariate adjustment, and can be extended to search for interactions. Here, we demonstrate the power and empirical type-I error of our approach using simulated case-control data sets of up to 500 K SNPs, a real genome-wide data set of 300 K SNPs, and a sequence-based dataset, each of which can be analysed in a few hours on a desktop workstation.
一次检测一个单核苷酸多态性(SNP)并不能充分发挥全基因组关联研究的潜力,以识别多个致病变异,而对于许多复杂疾病来说,这是一种合理的情况。我们表明,由于随机搜索方法的发展,现在可以对全基因组研究中的所有SNP进行同时分析,以识别最能预测疾病结局的子集。我们使用了一种受贝叶斯启发的惩罚最大似然方法,其中每个SNP都可以考虑对疾病风险的加性、显性和隐性贡献。获得了回归系数的后验模式估计值,每个估计值都被赋予了一个在零处有尖锐模式的先验。非零系数估计值被解释为对应于一个显著的SNP。我们研究了两种先验分布,并表明与单SNP检验相比,正态-指数-伽马先验导致了更好的SNP选择。我们还推导了一个明确的I型错误近似值,避免了使用置换程序的需要。除了全基因组分析外,我们的方法非常适合于对通过重测序和/或推断获得的非常密集的SNP集进行精细定位。它可以适应定量以及病例对照表型、协变量调整,并且可以扩展到搜索相互作用。在这里,我们使用多达50万个SNP的模拟病例对照数据集、一个30万个SNP的真实全基因组数据集和一个基于序列的数据集来证明我们方法的功效和经验性I型错误,每个数据集在台式工作站上只需几个小时就能进行分析。