Liu Jiangang, Campen Andrew, Huang Shuguang, Peng Sheng-Bin, Ye Xiang, Palakal Mathew, Dunker A Keith, Xia Yuni, Li Shuyu
Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN 46285, USA.
BMC Med Genomics. 2008 Sep 11;1:39. doi: 10.1186/1755-8794-1-39.
Numerous studies have used microarrays to identify gene signatures for predicting cancer patient clinical outcome and responses to chemotherapy. However, the potential impact of gene expression profiling in cancer diagnosis, prognosis and development of personalized treatment may not be fully exploited due to the lack of consensus gene signatures and poor understanding of the underlying molecular mechanisms.
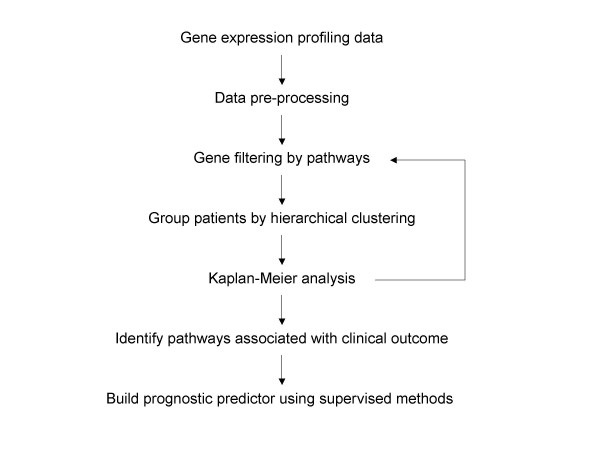
We developed a novel approach to derive gene signatures for breast cancer prognosis in the context of known biological pathways. Using unsupervised methods, cancer patients were separated into distinct groups based on gene expression patterns in one of the following pathways: apoptosis, cell cycle, angiogenesis, metastasis, p53, DNA repair, and several receptor-mediated signaling pathways including chemokines, EGF, FGF, HIF, MAP kinase, JAK and NF-kappaB. The survival probabilities were then compared between the patient groups to determine if differential gene expression in a specific pathway is correlated with differential survival.
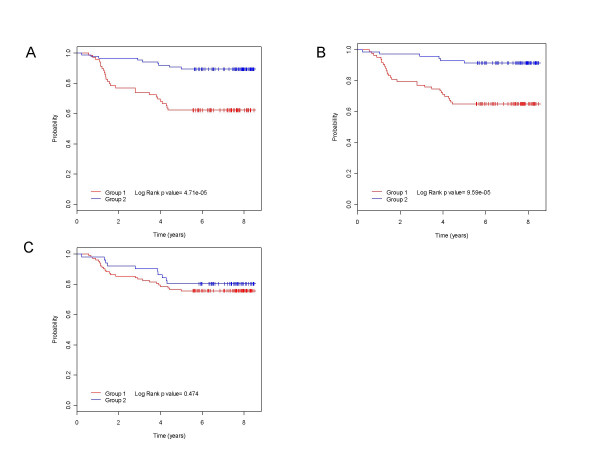
Our results revealed expression of cell cycle genes is strongly predictive of breast cancer outcomes. We further confirmed this observation by building a cell cycle gene signature model using supervised methods. Validated in multiple independent datasets, the cell cycle gene signature is a more accurate predictor for breast cancer clinical outcome than the previously identified Amsterdam 70-gene signature that has been developed into a FDA approved clinical test MammaPrint.
Taken together, the gene expression signature model we developed from well defined pathways is not only a consistently powerful prognosticator but also mechanistically linked to cancer biology. Our approach provides an alternative to the current methodology of identifying gene expression markers for cancer prognosis and drug responses using the whole genome gene expression data.
众多研究已使用微阵列来识别预测癌症患者临床结局及化疗反应的基因特征。然而,由于缺乏一致的基因特征且对潜在分子机制理解不足,基因表达谱在癌症诊断、预后及个性化治疗发展中的潜在影响可能未得到充分利用。
我们开发了一种新方法,在已知生物学途径的背景下推导乳腺癌预后的基因特征。使用无监督方法,根据以下途径之一中的基因表达模式将癌症患者分为不同组:细胞凋亡、细胞周期、血管生成、转移、p53、DNA修复,以及包括趋化因子、表皮生长因子(EGF)、成纤维细胞生长因子(FGF)、缺氧诱导因子(HIF)、丝裂原活化蛋白激酶(MAP激酶)、Janus激酶(JAK)和核因子κB(NF-κB)在内的几种受体介导的信号通路。然后比较患者组之间的生存概率,以确定特定途径中的差异基因表达是否与差异生存相关。
我们的结果显示细胞周期基因的表达强烈预测乳腺癌结局。我们通过使用监督方法构建细胞周期基因特征模型进一步证实了这一观察结果。在多个独立数据集中得到验证,与先前确定的已发展成为美国食品药品监督管理局(FDA)批准的临床检测方法MammaPrint的阿姆斯特丹70基因特征相比,细胞周期基因特征是乳腺癌临床结局更准确的预测指标。
综上所述,我们从明确界定的途径开发的基因表达特征模型不仅是一个始终强大的预后指标,而且在机制上与癌症生物学相关。我们的方法为目前使用全基因组基因表达数据识别癌症预后和药物反应的基因表达标志物的方法提供了一种替代方案。