Radulescu Ovidiu, Gorban Alexander N, Zinovyev Andrei, Lilienbaum Alain
IRMAR (CNRS UMR 6025), Université de Rennes 1, Rennes, France.
BMC Syst Biol. 2008 Oct 14;2:86. doi: 10.1186/1752-0509-2-86.
Cellular processes such as metabolism, decision making in development and differentiation, signalling, etc., can be modeled as large networks of biochemical reactions. In order to understand the functioning of these systems, there is a strong need for general model reduction techniques allowing to simplify models without loosing their main properties. In systems biology we also need to compare models or to couple them as parts of larger models. In these situations reduction to a common level of complexity is needed.
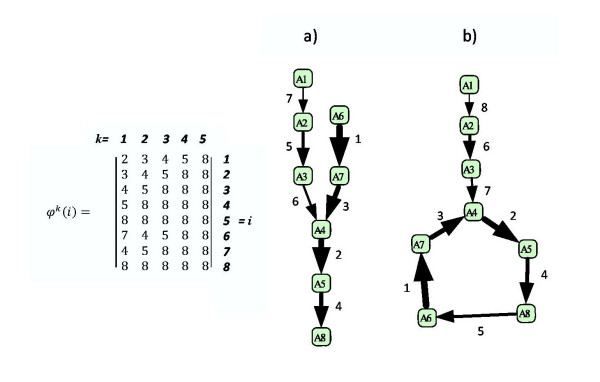
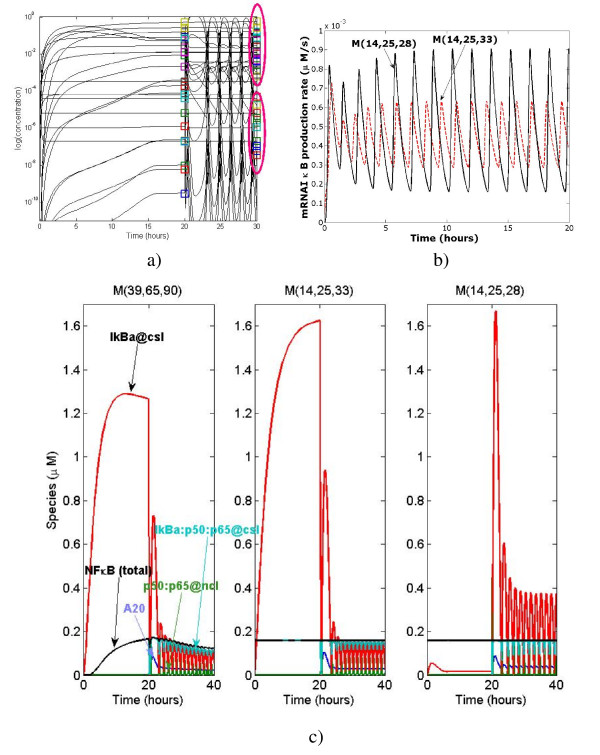
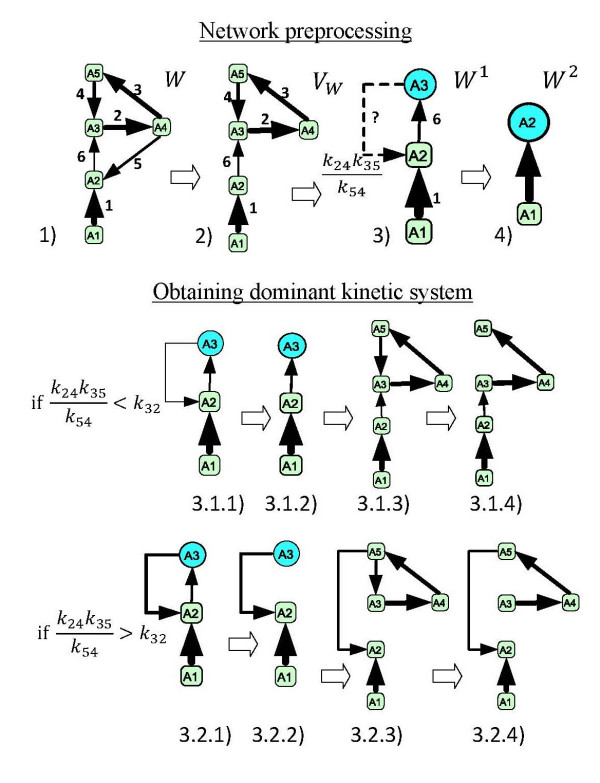
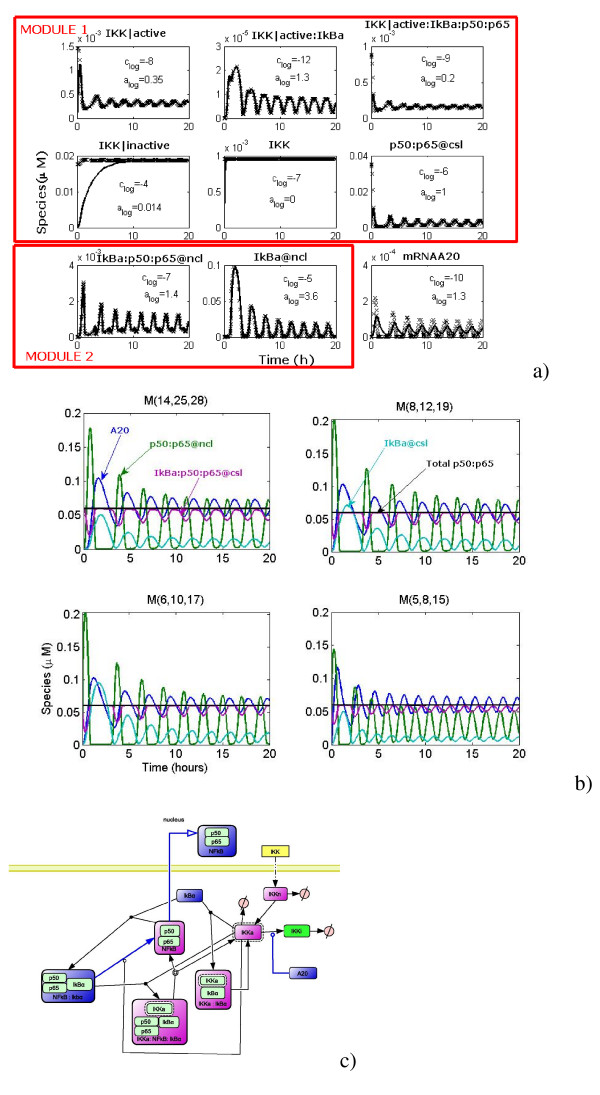
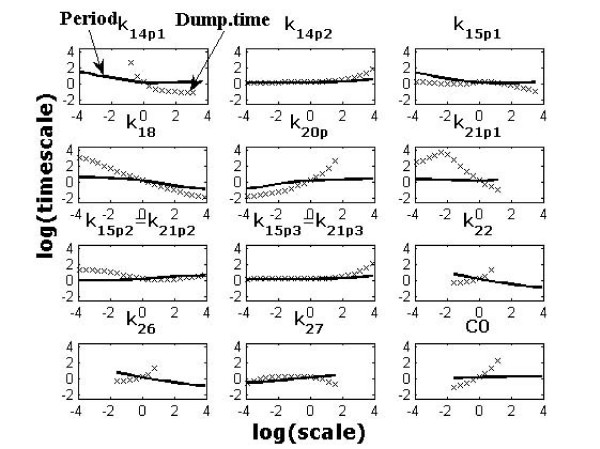
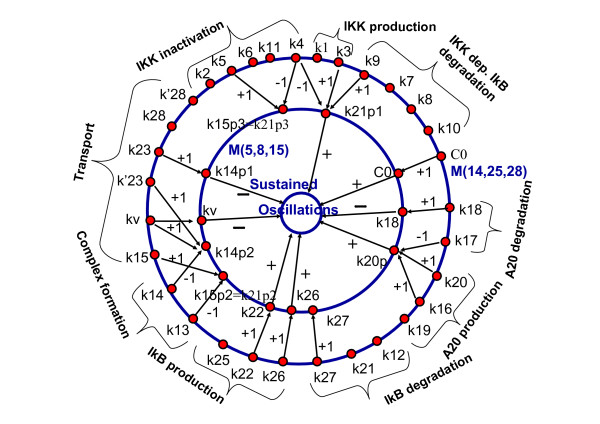
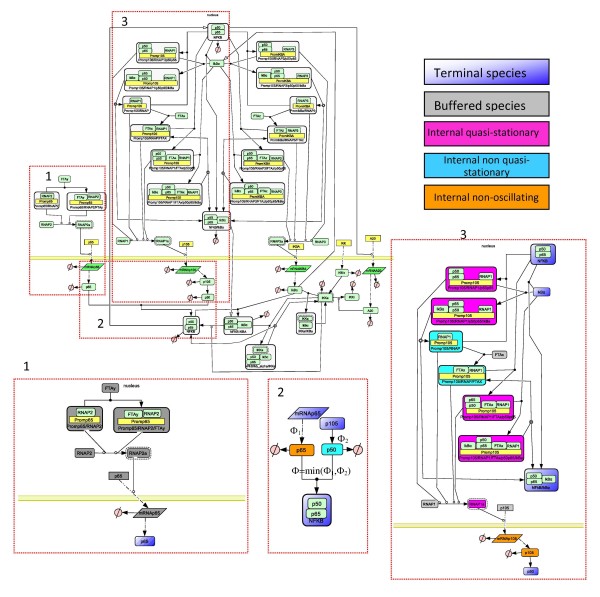
We propose a systematic treatment of model reduction of multiscale biochemical networks. First, we consider linear kinetic models, which appear as "pseudo-monomolecular" subsystems of multiscale nonlinear reaction networks. For such linear models, we propose a reduction algorithm which is based on a generalized theory of the limiting step that we have developed in 1. Second, for non-linear systems we develop an algorithm based on dominant solutions of quasi-stationarity equations. For oscillating systems, quasi-stationarity and averaging are combined to eliminate time scales much faster and much slower than the period of the oscillations. In all cases, we obtain robust simplifications and also identify the critical parameters of the model. The methods are demonstrated for simple examples and for a more complex model of NF-kappaB pathway.
Our approach allows critical parameter identification and produces hierarchies of models. Hierarchical modeling is important in "middle-out" approaches when there is need to zoom in and out several levels of complexity. Critical parameter identification is an important issue in systems biology with potential applications to biological control and therapeutics. Our approach also deals naturally with the presence of multiple time scales, which is a general property of systems biology models.
诸如新陈代谢、发育与分化中的决策制定、信号传导等细胞过程,可以被建模为大型生化反应网络。为了理解这些系统的功能,迫切需要通用的模型简化技术,以便在不丧失其主要特性的情况下简化模型。在系统生物学中,我们还需要比较模型或将它们作为更大模型的一部分进行耦合。在这些情况下,需要将模型简化到一个共同的复杂程度。
我们提出了一种对多尺度生化网络模型简化的系统处理方法。首先,我们考虑线性动力学模型,它们表现为多尺度非线性反应网络的“伪单分子”子系统。对于此类线性模型,我们提出了一种基于我们在文献[1]中所发展的极限步骤广义理论的简化算法。其次,对于非线性系统,我们开发了一种基于准稳态方程主导解的算法。对于振荡系统,将准稳态和平均相结合,以消除比振荡周期快得多和慢得多的时间尺度。在所有情况下,我们都获得了稳健的简化结果,并确定了模型的关键参数。通过简单示例和更复杂的NF-κB信号通路模型对这些方法进行了演示。
我们的方法能够识别关键参数并生成模型层次结构。在需要在几个复杂程度级别之间进行放大和缩小的“由中向外”方法中,层次建模很重要。关键参数识别是系统生物学中的一个重要问题,在生物控制和治疗方面具有潜在应用。我们的方法还自然地处理了多时间尺度的存在,这是系统生物学模型的一个普遍特性。