Cubillos-Ruiz Andrés, Morales Juan, Zambrano María Mercedes
Corporación Corpogen, Carrera 5 # 66A-34, Bogotá, Colombia.
BMC Res Notes. 2008 Nov 7;1:110. doi: 10.1186/1756-0500-1-110.
The recent determination of the complete nucleotide sequence of several Mycobacterium tuberculosis (MTB) genomes allows the use of comparative genomics as a tool for dissecting the nature and consequence of genetic variability within this species. The multiple alignment of the genomes of clinical strains (CDC1551, F11, Haarlem and C), along with the genomes of laboratory strains (H37Rv and H37Ra), provides new insights on the mechanisms of adaptation of this bacterium to the human host.
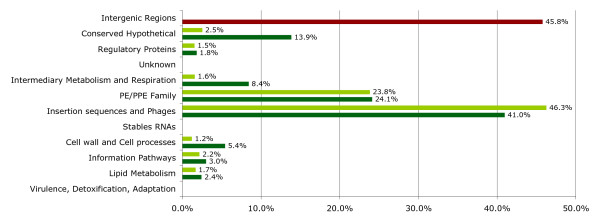
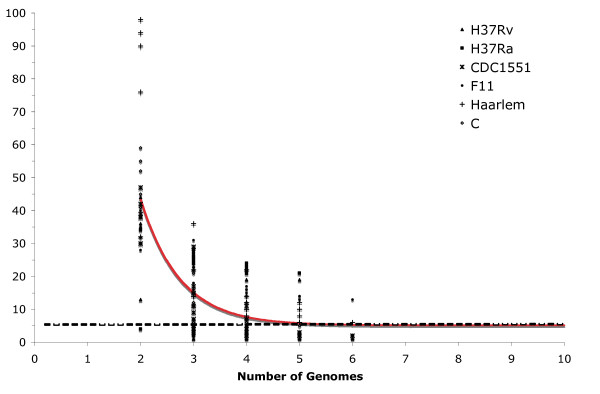
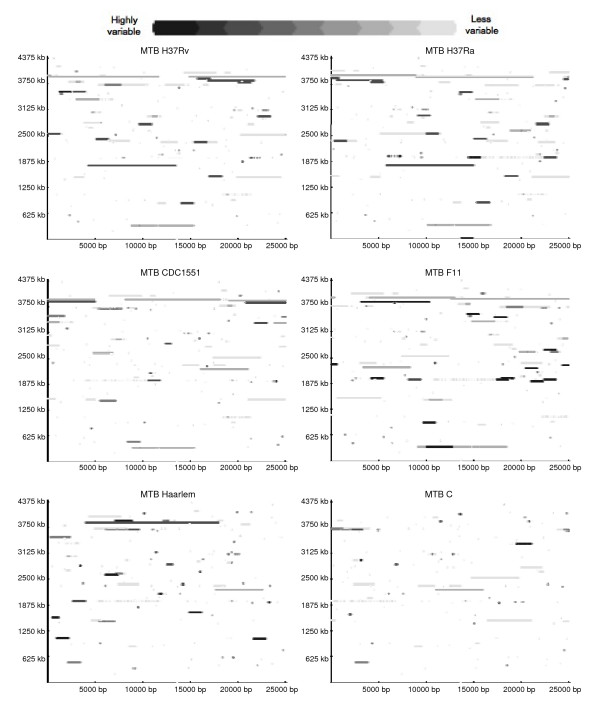
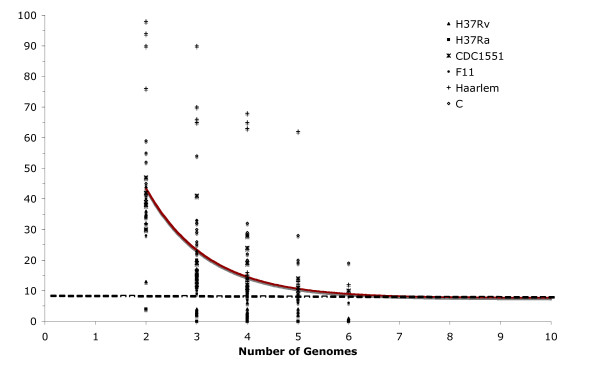
The genetic variation found in six M. tuberculosis strains does not involve significant genomic rearrangements. Most of the variation results from deletion and transposition events preferentially associated with insertion sequences and genes of the PE/PPE family but not with genes implicated in virulence. Using a Perl-based software islandsanalyser, which creates a representation of the genetic variation in the genome, we identified differences in the patterns of distribution and frequency of the polymorphisms across the genome. The identification of genes displaying strain-specific polymorphisms and the extrapolation of the number of strain-specific polymorphisms to an unlimited number of genomes indicates that the different strains contain a limited number of unique polymorphisms.
The comparison of multiple genomes demonstrates that the M. tuberculosis genome is currently undergoing an active process of gene decay, analogous to the adaptation process of obligate bacterial symbionts. This observation opens new perspectives into the evolution and the understanding of the pathogenesis of this bacterium.
最近对多个结核分枝杆菌(MTB)基因组完整核苷酸序列的测定,使得比较基因组学可作为剖析该物种内遗传变异性的本质及后果的工具。临床菌株(CDC1551、F11、哈勒姆株和C株)以及实验室菌株(H37Rv和H37Ra)基因组的多重比对,为该细菌适应人类宿主的机制提供了新见解。
在六种结核分枝杆菌菌株中发现的遗传变异并不涉及显著的基因组重排。大多数变异源于缺失和转座事件,这些事件优先与插入序列以及PE/PPE家族的基因相关,而非与毒力相关基因有关。使用基于Perl的软件islandsanalyser(该软件可呈现基因组中的遗传变异),我们确定了全基因组中多态性的分布模式和频率差异。对显示菌株特异性多态性的基因的鉴定,以及将菌株特异性多态性的数量外推至无限数量的基因组,表明不同菌株包含有限数量的独特多态性。
多个基因组的比较表明,结核分枝杆菌基因组目前正经历一个活跃的基因衰减过程,类似于专性细菌共生体的适应过程。这一观察结果为该细菌的进化及发病机制的理解开辟了新视角。