Key Laboratory of Symbol Computation and Knowledge Engineering of the Ministry of Education, College of Computer Science and Technology, Jilin University, Changchun 130012, China ; College of Chemistry, Jilin University, Changchun 130012, China.
Biomed Res Int. 2013;2013:409062. doi: 10.1155/2013/409062. Epub 2013 Aug 29.
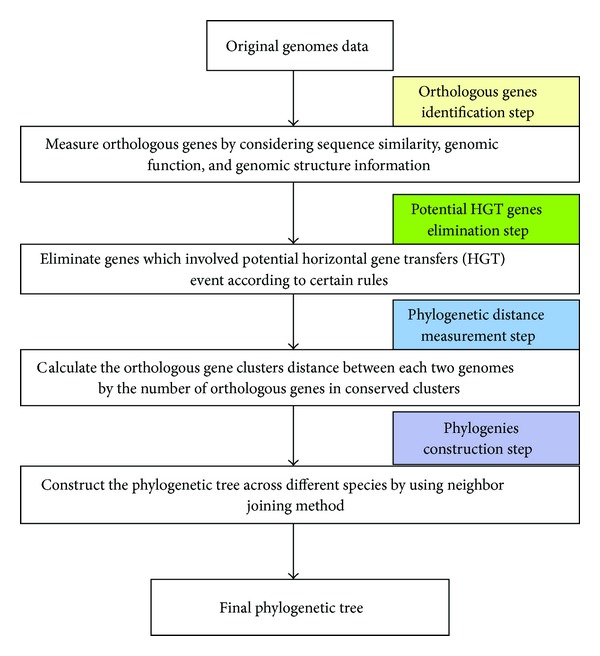
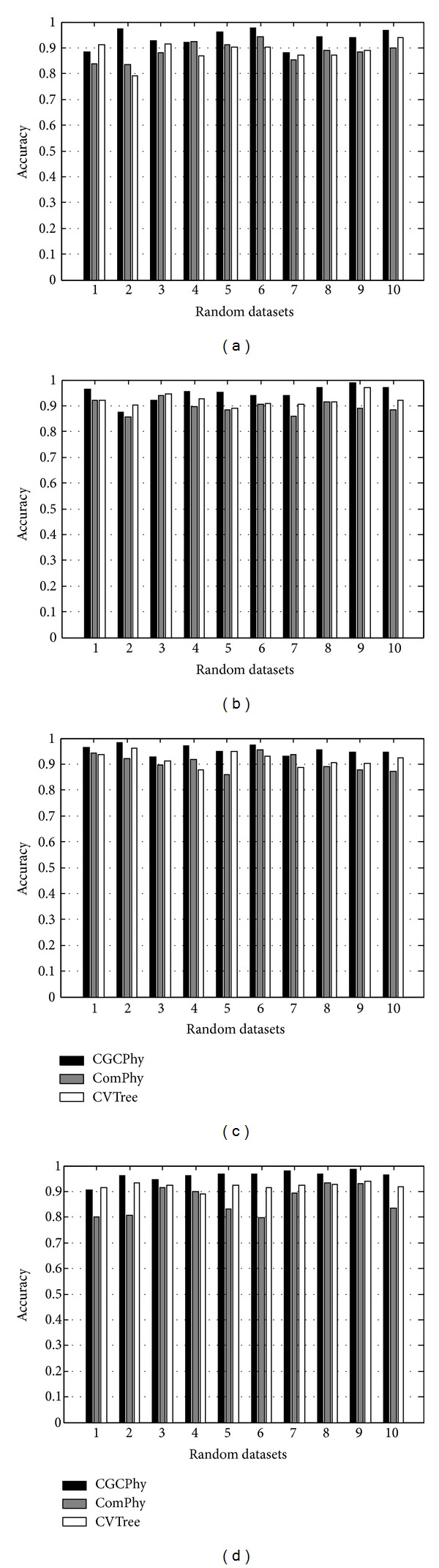
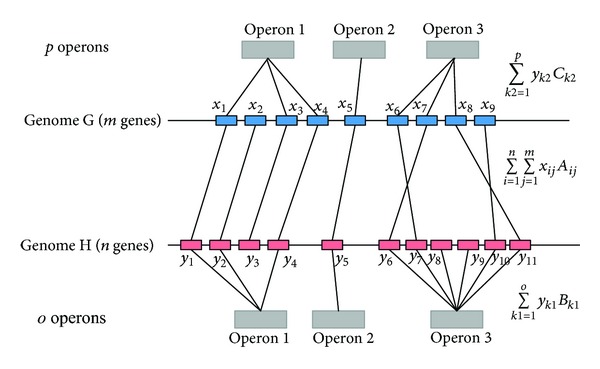
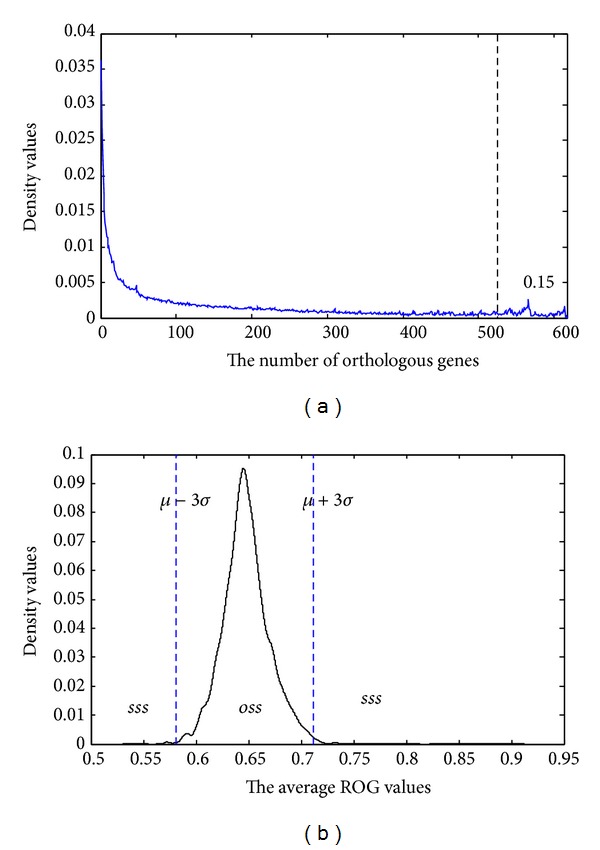
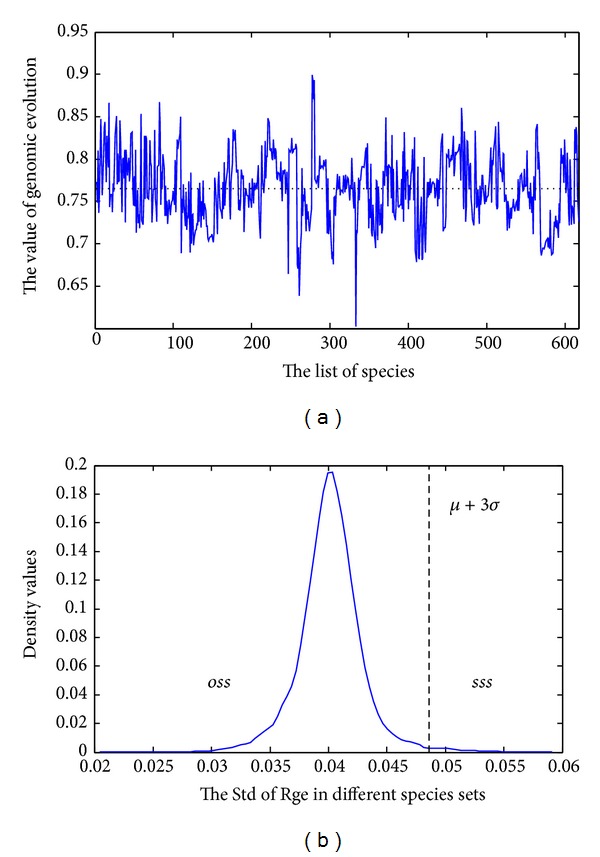
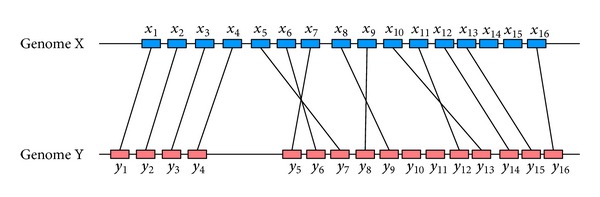
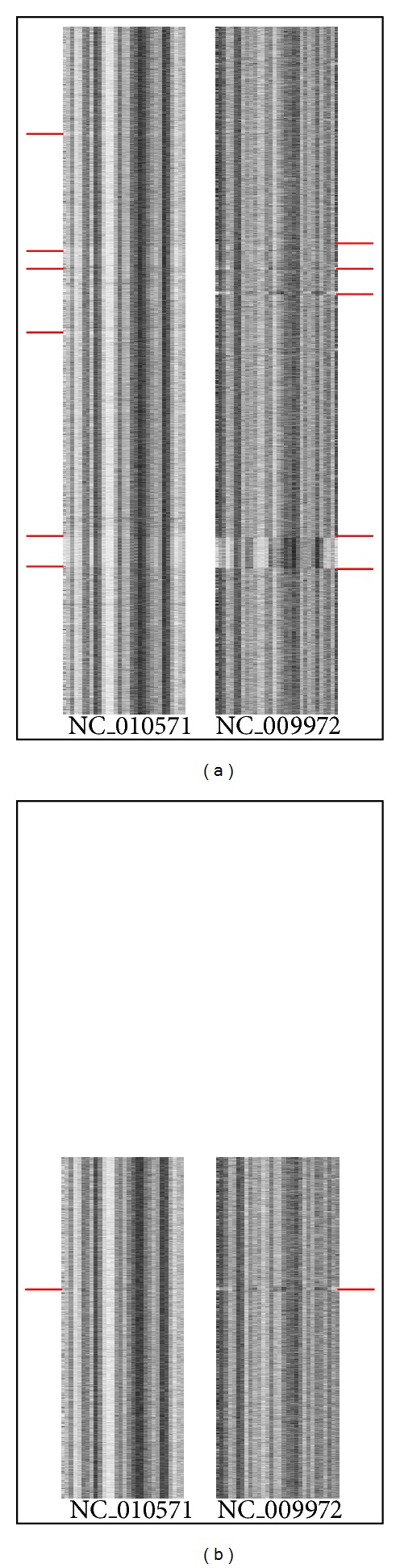
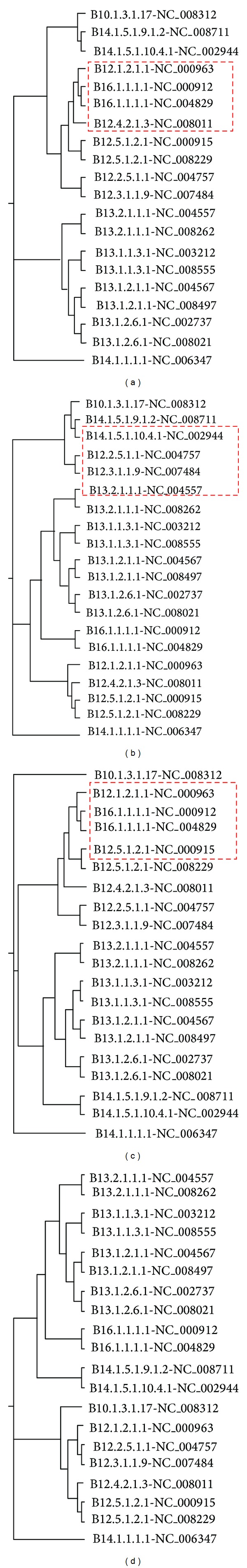
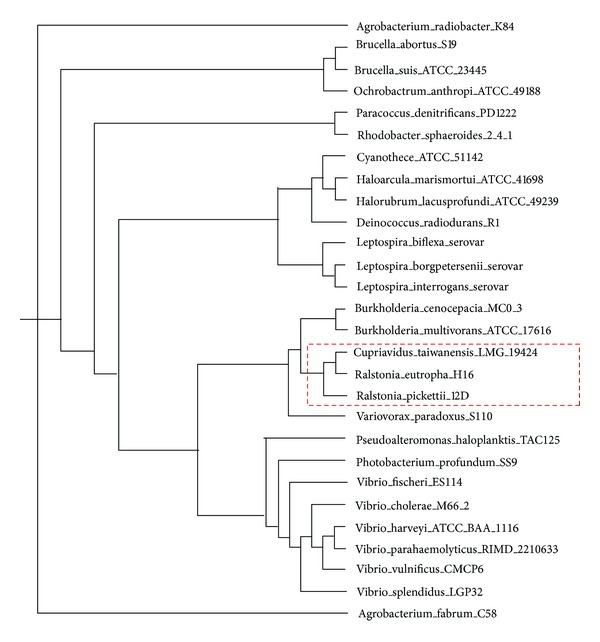
Phylogenetic trees are used to represent the evolutionary relationship among various groups of species. In this paper, a novel method for inferring prokaryotic phylogenies using multiple genomic information is proposed. The method is called CGCPhy and based on the distance matrix of orthologous gene clusters between whole-genome pairs. CGCPhy comprises four main steps. First, orthologous genes are determined by sequence similarity, genomic function, and genomic structure information. Second, genes involving potential HGT events are eliminated, since such genes are considered to be the highly conserved genes across different species and the genes located on fragments with abnormal genome barcode. Third, we calculate the distance of the orthologous gene clusters between each genome pair in terms of the number of orthologous genes in conserved clusters. Finally, the neighbor-joining method is employed to construct phylogenetic trees across different species. CGCPhy has been examined on different datasets from 617 complete single-chromosome prokaryotic genomes and achieved applicative accuracies on different species sets in agreement with Bergey's taxonomy in quartet topologies. Simulation results show that CGCPhy achieves high average accuracy and has a low standard deviation on different datasets, so it has an applicative potential for phylogenetic analysis.
系统发育树用于表示不同物种群体之间的进化关系。本文提出了一种使用多种基因组信息推断原核生物系统发育的新方法。该方法称为 CGCPhy,基于全基因组对之间的直系同源基因簇的距离矩阵。CGCPhy 包括四个主要步骤。首先,通过序列相似性、基因组功能和基因组结构信息确定直系同源基因。其次,消除涉及潜在 HGT 事件的基因,因为这些基因被认为是不同物种之间高度保守的基因,并且位于具有异常基因组条码的片段上的基因。第三,我们根据保守簇中直系同源基因的数量计算每个基因组对之间直系同源基因簇的距离。最后,采用邻接法构建不同物种之间的系统发育树。CGCPhy 已在来自 617 个完整单染色体原核生物基因组的不同数据集上进行了检查,并在与 Bergey 的分类学一致的四联体拓扑中,在不同的物种集上实现了适用的准确性。模拟结果表明,CGCPhy 在不同数据集上具有较高的平均准确性和较低的标准偏差,因此在系统发育分析方面具有应用潜力。