Costa Ivan G, Schönhuth Alexander, Hafemeister Christoph, Schliep Alexander
Center of Informatics, Federal University of Pernambuco, Recife, Brazil.
Bioinformatics. 2009 Jun 15;25(12):i6-14. doi: 10.1093/bioinformatics/btp222.
Personalized medicine based on molecular aspects of diseases, such as gene expression profiling, has become increasingly popular. However, one faces multiple challenges when analyzing clinical gene expression data; most of the well-known theoretical issues such as high dimension of feature spaces versus few examples, noise and missing data apply. Special care is needed when designing classification procedures that support personalized diagnosis and choice of treatment. Here, we particularly focus on classification of interferon-beta (IFNbeta) treatment response in Multiple Sclerosis (MS) patients which has attracted substantial attention in the recent past. Half of the patients remain unaffected by IFNbeta treatment, which is still the standard. For them the treatment should be timely ceased to mitigate the side effects.
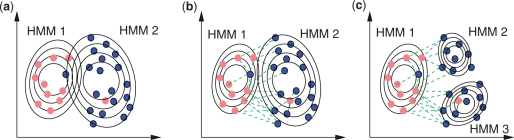
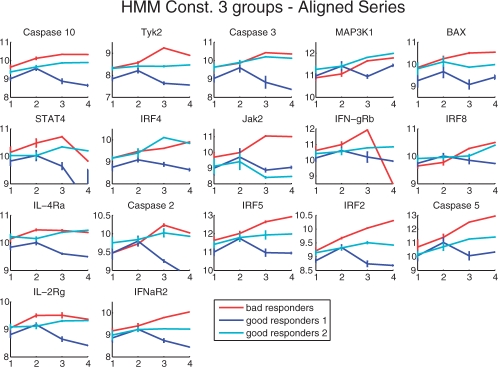
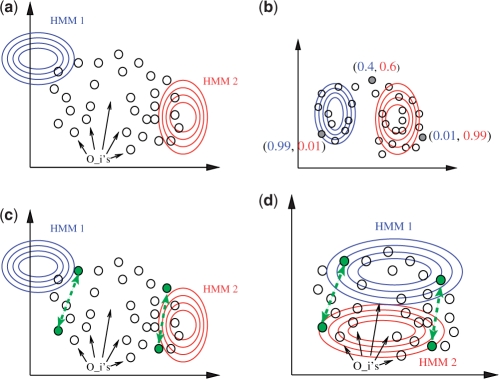
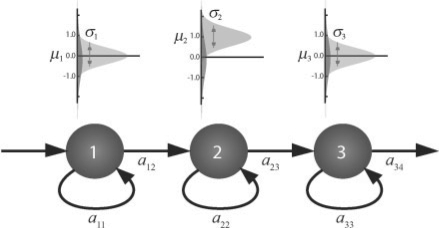
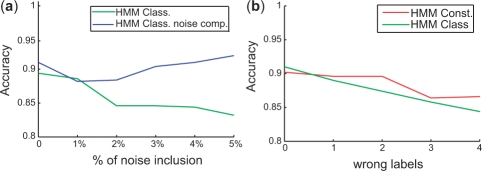
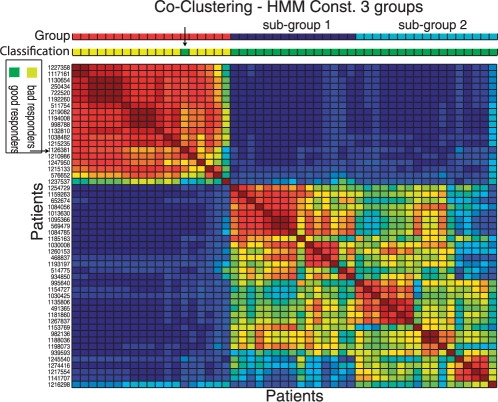
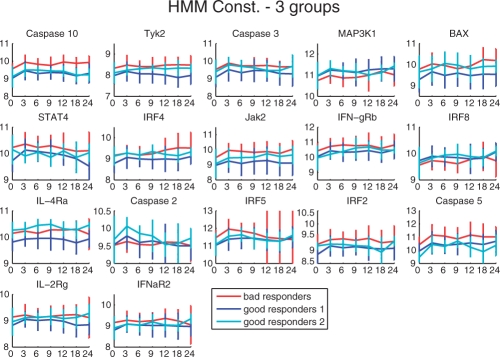
We propose constrained estimation of mixtures of hidden Markov models as a methodology to classify patient response to IFNbeta treatment. The advantages of our approach are that it takes the temporal nature of the data into account and its robustness with respect to noise, missing data and mislabeled samples. Moreover, mixture estimation enables to explore the presence of response sub-groups of patients on the transcriptional level. We clearly outperformed all prior approaches in terms of prediction accuracy, raising it, for the first time, >90%. Additionally, we were able to identify potentially mislabeled samples and to sub-divide the good responders into two sub-groups that exhibited different transcriptional response programs. This is supported by recent findings on MS pathology and therefore may raise interesting clinical follow-up questions.
The method is implemented in the GQL framework and is available at http://www.ghmm.org/gql. Datasets are available at http://www.cin.ufpe.br/ approximately igcf/MSConst.
Supplementary data are available at Bioinformatics online.
基于疾病分子特征的个性化医疗,如基因表达谱分析,已越来越受欢迎。然而,在分析临床基因表达数据时会面临多重挑战;大多数众所周知的理论问题,如特征空间维度高而样本少、噪声和缺失数据等都存在。在设计支持个性化诊断和治疗选择的分类程序时需要特别小心。在此,我们特别关注多发性硬化症(MS)患者中干扰素-β(IFNβ)治疗反应的分类,这在最近引起了广泛关注。仍有一半的患者对IFNβ治疗无反应,而IFNβ治疗仍是标准治疗方法。对于这些患者,应及时停止治疗以减轻副作用。
我们提出将隐藏马尔可夫模型混合的约束估计作为一种对患者对IFNβ治疗的反应进行分类的方法。我们方法的优点在于它考虑了数据的时间特性,并且对噪声、缺失数据和错误标记的样本具有鲁棒性。此外,混合估计能够在转录水平上探索患者反应亚组的存在。在预测准确性方面,我们明显优于所有先前的方法,首次将其提高到>90%。此外,我们能够识别潜在错误标记的样本,并将良好反应者细分为两个表现出不同转录反应程序的亚组。这得到了最近关于MS病理学研究结果的支持,因此可能会引发有趣的临床随访问题。
该方法在GQL框架中实现,可在http://www.ghmm.org/gql获取。数据集可在http://www.cin.ufpe.br/ approximately igcf/MSConst获取。
补充数据可在《生物信息学》在线版获取。