Reid John E, Ott Sascha, Wernisch Lorenz
MRC Biostatistics Unit, Institute of Public Health, University Forvie Site, Cambridge CB2 0SR, UK.
BMC Bioinformatics. 2009 Jul 16;10:218. doi: 10.1186/1471-2105-10-218.
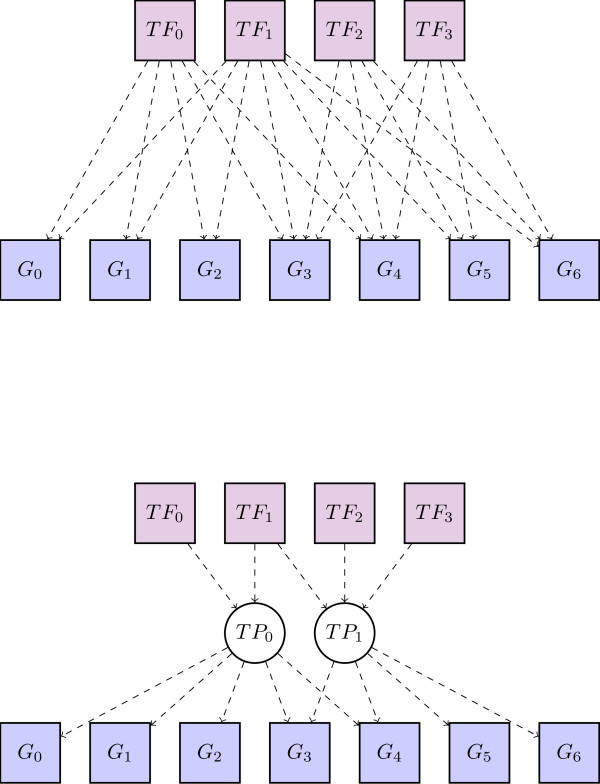
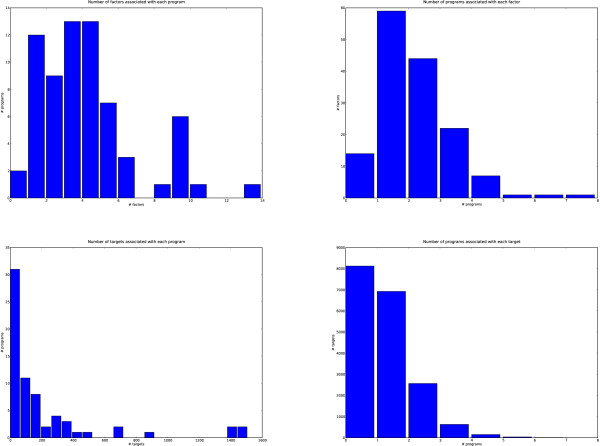
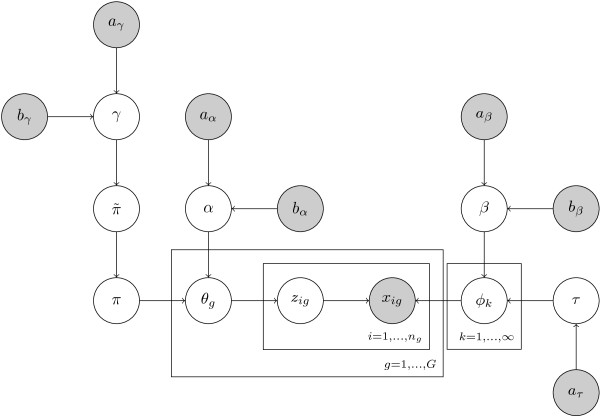
Transcriptional regulation is an important part of regulatory control in eukaryotes. Even if binding motifs for transcription factors are known, the task of finding binding sites by scanning sequences is plagued by false positives. One way to improve the detection of binding sites from motifs is by taking cooperativity of transcription factor binding into account. We propose a non-parametric probabilistic model, similar to a document topic model, for detecting transcriptional programs, groups of cooperative transcription factors and co-regulated genes. The analysis results in transcriptional programs which generalise both transcriptional modules and TF-target gene incidence matrices and provide a higher-level summary of these structures. The method is independent of prior specification of training sets of genes, for example, via gene expression data. The analysis is based on known binding motifs.
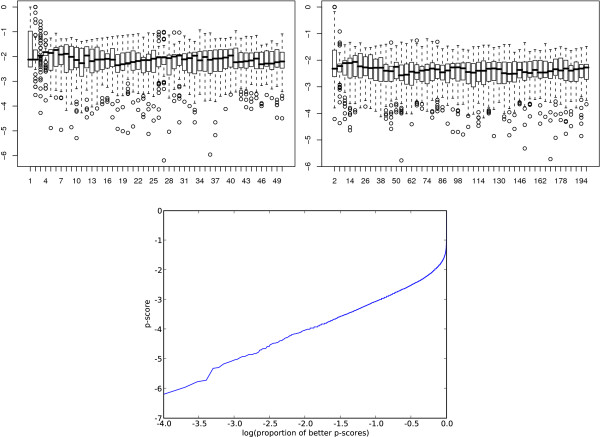
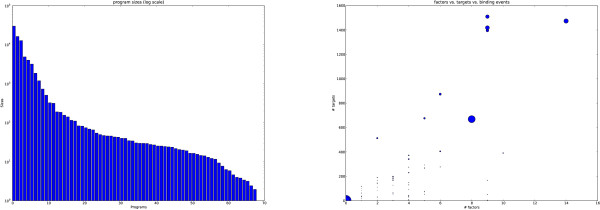
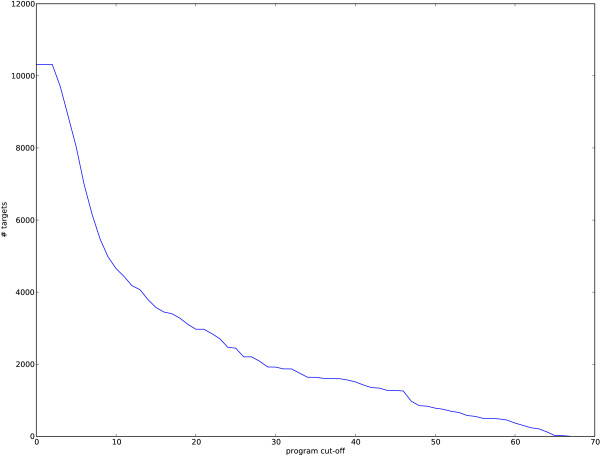
We applied our method to putative regulatory regions of 18,445 Mus musculus genes. We discovered just 68 transcriptional programs that effectively summarised the action of 149 transcription factors on these genes. Several of these programs were significantly enriched for known biological processes and signalling pathways. One transcriptional program has a significant overlap with a reference set of cell cycle specific transcription factors.
Our method is able to pick out higher order structure from noisy sequence analyses. The transcriptional programs it identifies potentially represent common mechanisms of regulatory control across the genome. It simultaneously predicts which genes are co-regulated and which sets of transcription factors cooperate to achieve this co-regulation. The programs we discovered enable biologists to choose new genes and transcription factors to study in specific transcriptional regulatory systems.
转录调控是真核生物调控控制的重要组成部分。即使转录因子的结合基序已知,通过扫描序列来寻找结合位点的任务也会受到假阳性的困扰。从基序中提高结合位点检测的一种方法是考虑转录因子结合的协同性。我们提出了一种非参数概率模型,类似于文档主题模型,用于检测转录程序、协同转录因子组和共调控基因。分析结果得到的转录程序概括了转录模块和转录因子-靶基因关联矩阵,并对这些结构提供了更高级别的总结。该方法独立于例如通过基因表达数据对基因训练集的先验指定。分析基于已知的结合基序。
我们将我们的方法应用于18445个小家鼠基因的假定调控区域。我们发现仅68个转录程序就有效地概括了149个转录因子对这些基因的作用。其中几个程序在已知的生物学过程和信号通路中显著富集。一个转录程序与一组细胞周期特异性转录因子的参考集有显著重叠。
我们的方法能够从嘈杂的序列分析中挑选出高阶结构。它识别出的转录程序可能代表了全基因组调控控制的共同机制。它同时预测哪些基因是共调控的,以及哪些转录因子组协同实现这种共调控。我们发现的程序使生物学家能够选择新的基因和转录因子,以便在特定的转录调控系统中进行研究。