Liu Xiangdong, Jessen Walter J, Sivaganesan Siva, Aronow Bruce J, Medvedovic Mario
Department of Environmental Health, University of Cincinnati, 3223 Eden Ave, ML 56, Cincinnati, Ohio 45267, USA.
BMC Bioinformatics. 2007 Aug 3;8:283. doi: 10.1186/1471-2105-8-283.
Transcriptional modules (TM) consist of groups of co-regulated genes and transcription factors (TF) regulating their expression. Two high-throughput (HT) experimental technologies, gene expression microarrays and Chromatin Immuno-Precipitation on Chip (ChIP-chip), are capable of producing data informative about expression regulatory mechanism on a genome scale. The optimal approach to joint modeling of data generated by these two complementary biological assays, with the goal of identifying and characterizing TMs, is an important open problem in computational biomedicine.
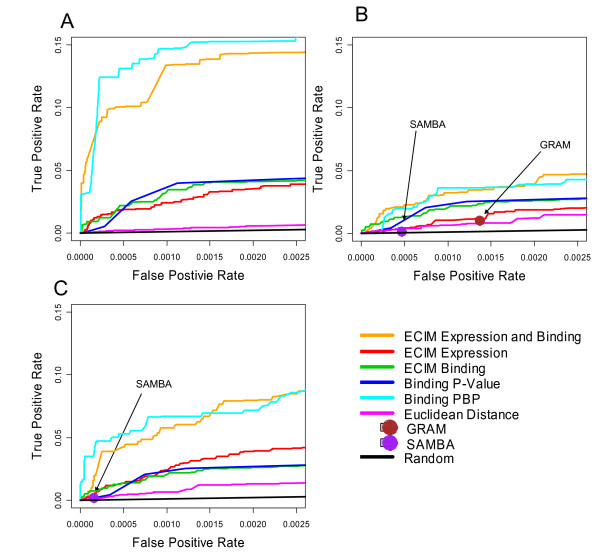
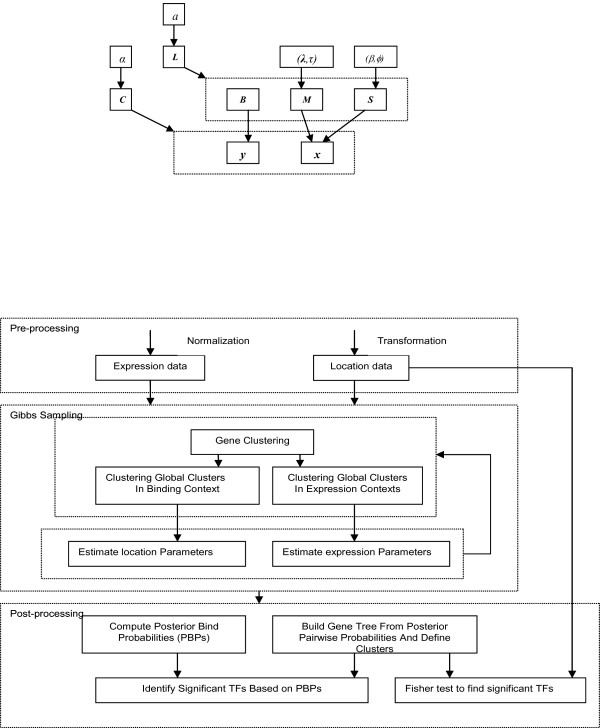
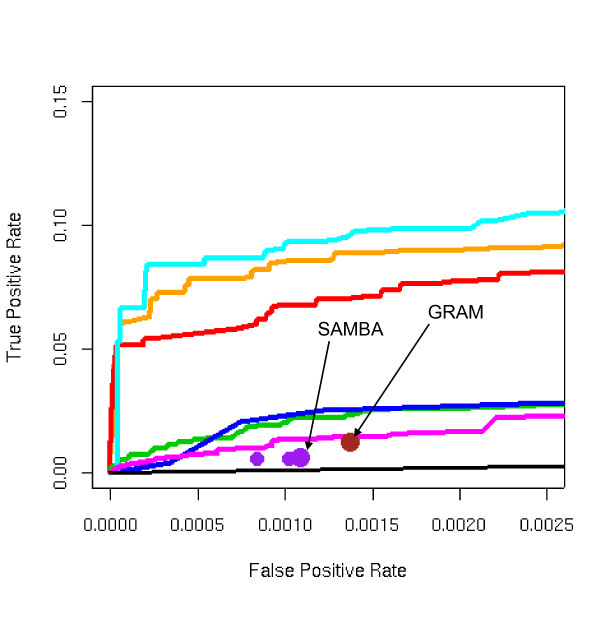
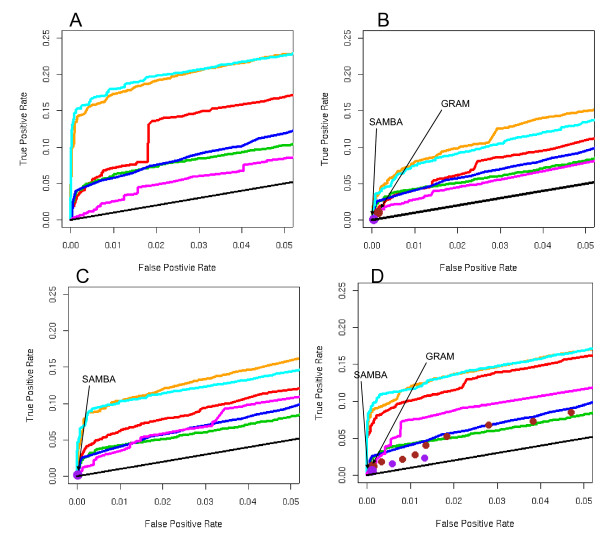
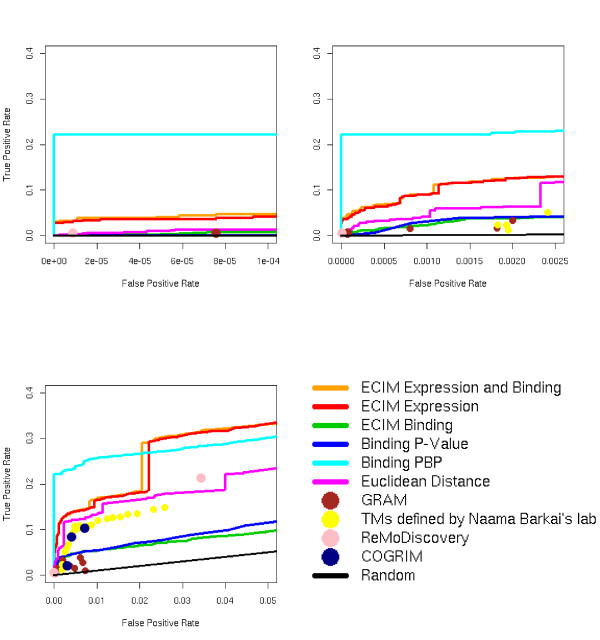
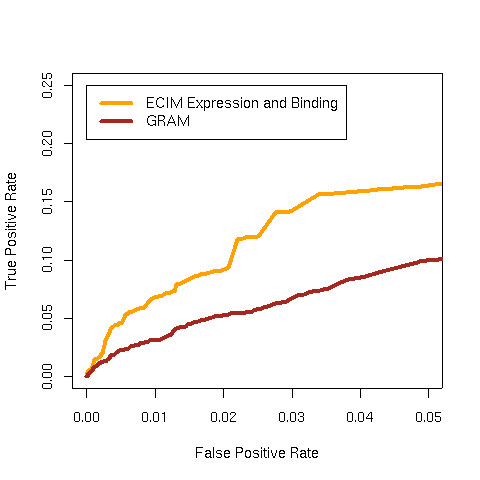
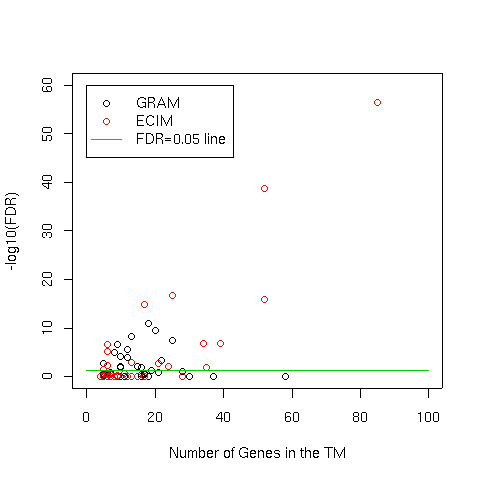
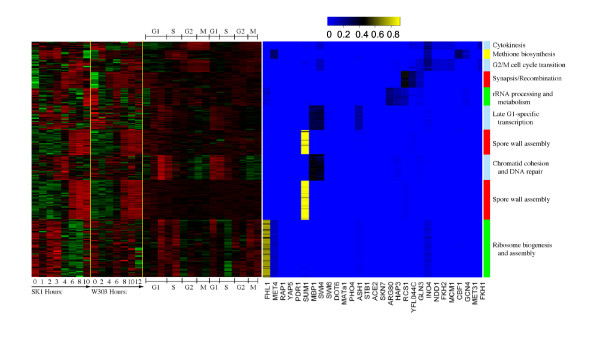
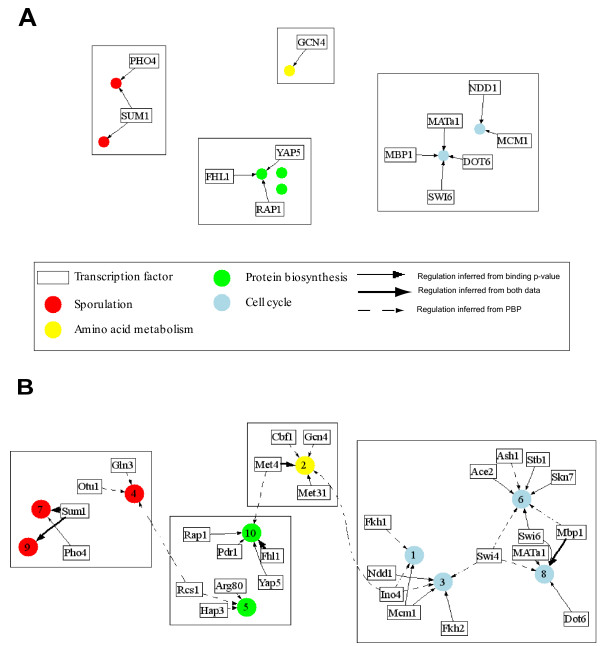
We developed and validated a novel probabilistic model and related computational procedure for identifying TMs by jointly modeling gene expression and ChIP-chip binding data. We demonstrate an improved functional coherence of the TMs produced by the new method when compared to either analyzing expression or ChIP-chip data separately or to alternative approaches for joint analysis. We also demonstrate the ability of the new algorithm to identify novel regulatory relationships not revealed by ChIP-chip data alone. The new computational procedure can be used in more or less the same way as one would use simple hierarchical clustering without performing any special transformation of data prior to the analysis. The R and C-source code for implementing our algorithm is incorporated within the R package gimmR which is freely available at http://eh3.uc.edu/gimm.
Our results indicate that, whenever available, ChIP-chip and expression data should be analyzed within the unified probabilistic modeling framework, which will likely result in improved clusters of co-regulated genes and improved ability to detect meaningful regulatory relationships. Given the good statistical properties and the ease of use, the new computational procedure offers a worthy new tool for reconstructing transcriptional regulatory networks.
转录模块(TM)由共同调控的基因群以及调控其表达的转录因子(TF)组成。两种高通量(HT)实验技术,即基因表达微阵列和芯片染色质免疫沉淀技术(ChIP-chip),能够在基因组规模上产生有关表达调控机制的信息丰富的数据。以识别和表征转录模块为目标,对这两种互补生物学检测产生的数据进行联合建模的最佳方法,是计算生物医学中一个重要的开放性问题。
我们开发并验证了一种新的概率模型及相关计算程序,通过对基因表达和ChIP-chip结合数据进行联合建模来识别转录模块。与单独分析表达数据或ChIP-chip数据,或与联合分析的其他方法相比,我们证明了新方法产生的转录模块具有更高的功能一致性。我们还展示了新算法识别仅ChIP-chip数据未揭示的新型调控关系的能力。新的计算程序使用方式与简单层次聚类大致相同,在分析前无需对数据进行任何特殊转换。实现我们算法的R和C源代码包含在R包gimmR中,可从http://eh3.uc.edu/gimm免费获取。
我们的结果表明,只要可行,应在统一的概率建模框架内分析ChIP-chip和表达数据,这可能会改善共调控基因的聚类,并提高检测有意义调控关系的能力。鉴于良好的统计特性和易用性,新的计算程序为重建转录调控网络提供了一个有价值的新工具。