Systems Biology Centre, University of Warwick, Coventry, CV4 7AL, UK.
Bioinformatics. 2010 Jun 15;26(12):i158-67. doi: 10.1093/bioinformatics/btq210.
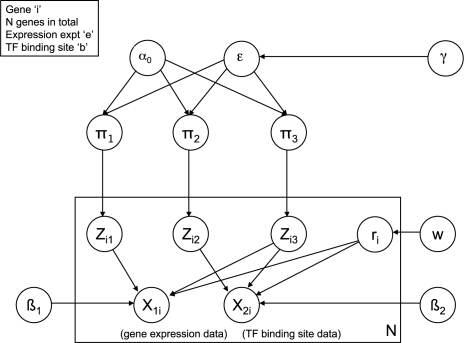
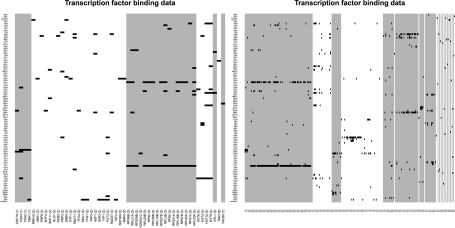
We present a method for directly inferring transcriptional modules (TMs) by integrating gene expression and transcription factor binding (ChIP-chip) data. Our model extends a hierarchical Dirichlet process mixture model to allow data fusion on a gene-by-gene basis. This encodes the intuition that co-expression and co-regulation are not necessarily equivalent and hence we do not expect all genes to group similarly in both datasets. In particular, it allows us to identify the subset of genes that share the same structure of transcriptional modules in both datasets.
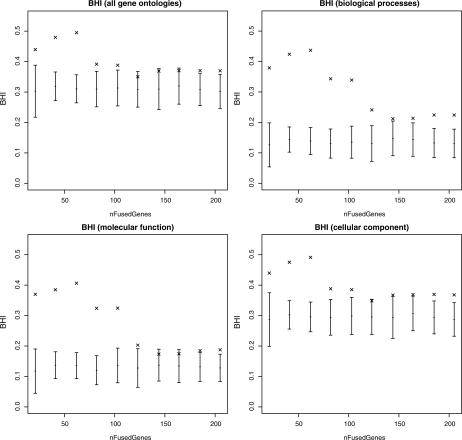
We find that by working on a gene-by-gene basis, our model is able to extract clusters with greater functional coherence than existing methods. By combining gene expression and transcription factor binding (ChIP-chip) data in this way, we are better able to determine the groups of genes that are most likely to represent underlying TMs.
If interested in the code for the work presented in this article, please contact the authors.
Supplementary data are available at Bioinformatics online.
我们提出了一种通过整合基因表达和转录因子结合(ChIP-chip)数据来直接推断转录模块(TMs)的方法。我们的模型扩展了层次狄利克雷过程混合模型,允许在逐基因的基础上进行数据融合。这就体现了这样一种直觉,即共表达和共调控不一定等同,因此我们不期望两个数据集的所有基因都以相似的方式分组。特别是,它允许我们确定在两个数据集中共有的转录模块结构的基因子集。
我们发现,通过逐基因的方式,我们的模型能够提取出比现有方法具有更高功能一致性的聚类。通过以这种方式结合基因表达和转录因子结合(ChIP-chip)数据,我们能够更好地确定最有可能代表潜在 TM 的基因组。
如果有兴趣了解本文中介绍的工作的代码,请联系作者。
补充数据可在《生物信息学》在线获取。