Istituto Tecnologie Biomediche, Consiglio Nazionale Ricerche, Via Fratelli Cervi 93, Segrate (MI), Italy.
BMC Bioinformatics. 2009 Oct 15;10 Suppl 12(Suppl 12):S8. doi: 10.1186/1471-2105-10-S12-S8.
The identification of the organisation and dynamics of molecular pathways is crucial for the understanding of cell function. In order to reconstruct the molecular pathways in which a gene of interest is involved in regulating a cell, it is important to identify the set of genes to which it interacts with to determine cell function. In this context, the mining and the integration of a large amount of publicly available data, regarding the transcriptome and the proteome states of a cell, are a useful resource to complement biological research.
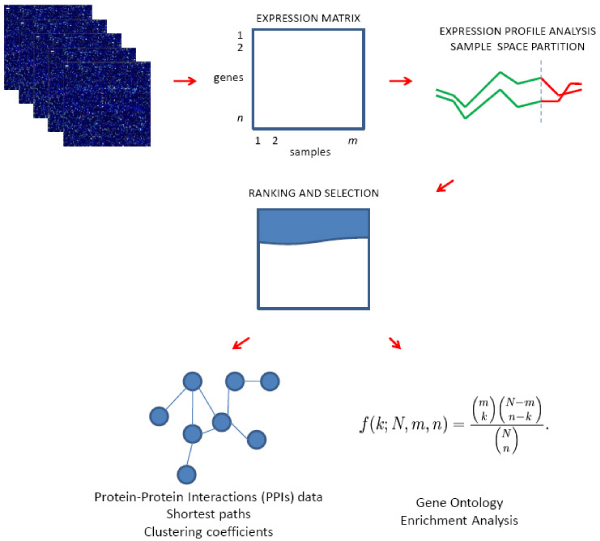
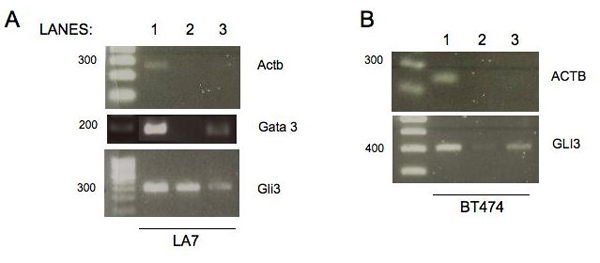
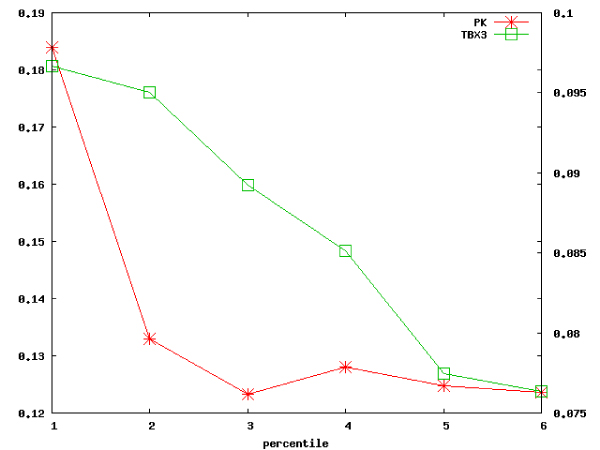
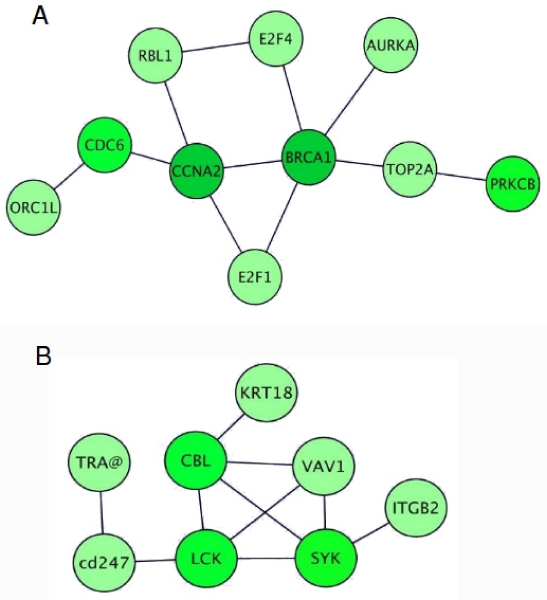
We describe an approach for the identification of genes that interact with each other to regulate cell function. The strategy relies on the analysis of gene expression profile similarity, considering large datasets of expression data. During the similarity evaluation, the methodology determines the most significant subset of samples in which the evaluated genes are highly correlated. Hence, the strategy enables the exclusion of samples that are not relevant for each gene pair analysed. This feature is important when considering a large set of samples characterised by heterogeneous experimental conditions where different pools of biological processes can be active across the samples. The putative partners of the studied gene are then further characterised, analysing the distribution of the Gene Ontology terms and integrating the protein-protein interaction (PPI) data. The strategy was applied for the analysis of the functional relationships of a gene of known function, Pyruvate Kinase, and for the prediction of functional partners of the human transcription factor TBX3. In both cases the analysis was done on a dataset composed by breast primary tumour expression data derived from the literature. Integration and analysis of PPI data confirmed the prediction of the methodology, since the genes identified to be functionally related were associated to proteins close in the PPI network. Two genes among the predicted putative partners of TBX3 (GLI3 and GATA3) were confirmed by in vivo binding assays (crosslinking immunoprecipitation, X-ChIP) in which the putative DNA enhancer sequence sites of GATA3 and GLI3 were found to be bound by the Tbx3 protein.
The presented strategy is demonstrated to be an effective approach to identify genes that establish functional relationships. The methodology identifies and characterises genes with a similar expression profile, through data mining and integrating data from publicly available resources, to contribute to a better understanding of gene regulation and cell function. The prediction of the TBX3 target genes GLI3 and GATA3 was experimentally confirmed.
鉴定分子途径的组成和动态对于理解细胞功能至关重要。为了重建感兴趣的基因参与调节细胞的分子途径,确定与它相互作用的一组基因以确定细胞功能非常重要。在这种情况下,挖掘和整合大量关于细胞转录组和蛋白质组状态的公开可用数据是补充生物学研究的有用资源。
我们描述了一种识别相互作用以调节细胞功能的基因的方法。该策略依赖于基因表达谱相似性的分析,考虑了大量的表达数据集。在相似性评估过程中,该方法确定了评估基因高度相关的最显著样本子集。因此,该策略能够排除与分析的每个基因对不相关的样本。当考虑一组由具有不同生物过程池在样本中活跃的异质实验条件所表征的大量样本时,此功能很重要。然后,进一步分析研究基因的假定伙伴,分析基因本体论术语的分布并整合蛋白质-蛋白质相互作用 (PPI) 数据。该策略用于分析已知功能基因丙酮酸激酶的功能关系,并预测人类转录因子 TBX3 的功能伙伴。在这两种情况下,分析都是在由文献中获得的乳腺原发性肿瘤表达数据组成的数据集上进行的。PPI 数据的集成和分析证实了该方法的预测,因为被鉴定为功能相关的基因与 PPI 网络中接近的蛋白质相关联。在 TBX3 的预测假定伙伴中,有两个基因(GLI3 和 GATA3)通过体内结合测定(交联免疫沉淀,X-ChIP)得到证实,其中发现 GATA3 和 GLI3 的假定 DNA 增强子序列位点被 Tbx3 蛋白结合。
所提出的策略被证明是一种识别建立功能关系的基因的有效方法。该方法通过数据挖掘和整合来自公开可用资源的数据,识别和表征具有相似表达谱的基因,有助于更好地理解基因调控和细胞功能。TBX3 靶基因 GLI3 和 GATA3 的预测得到了实验证实。