From the Departments of Oncological Sciences, University of Utah, Salt Lake City, Utah 84112; Departments of Huntsman Cancer Institute, University of Utah, Salt Lake City, Utah 84112 and; Departments of Howard Hughes Medical Institute, University of Utah, Salt Lake City, Utah 84112.
Departments of Huntsman Cancer Institute, University of Utah, Salt Lake City, Utah 84112 and.
J Biol Chem. 2010 Feb 5;285(6):4110-4121. doi: 10.1074/jbc.M109.073676. Epub 2009 Nov 29.
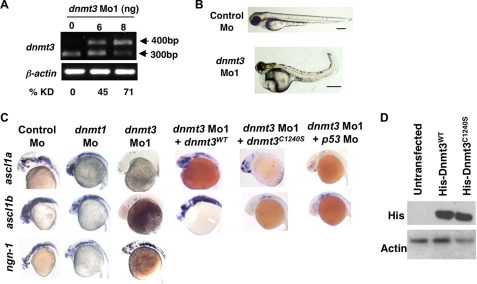
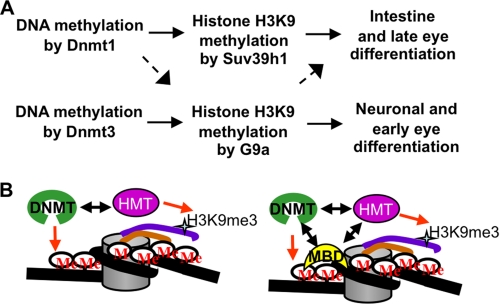
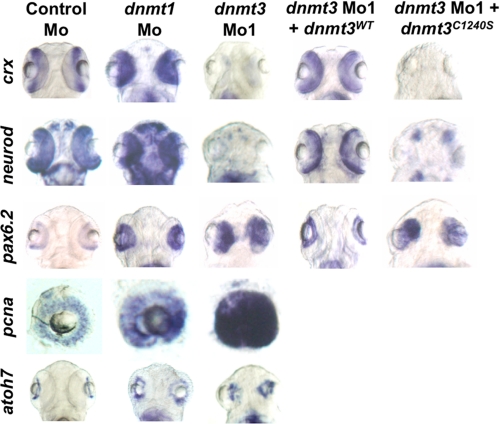
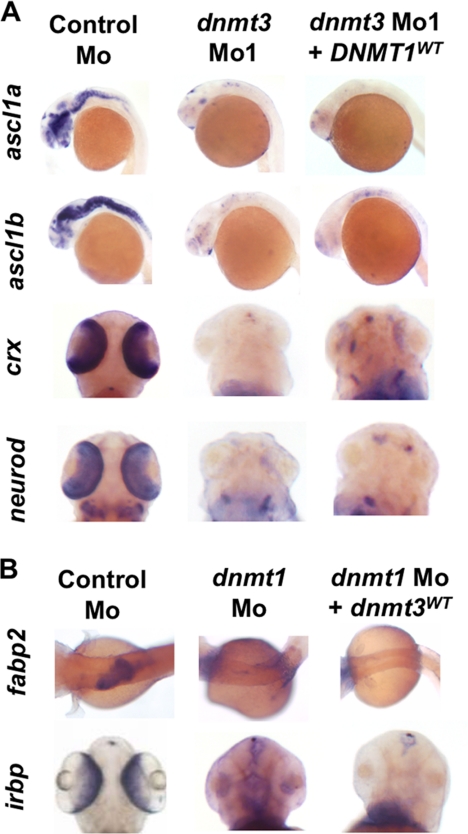
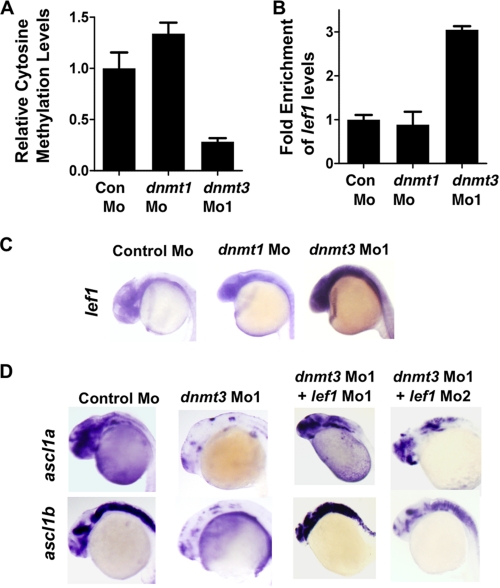
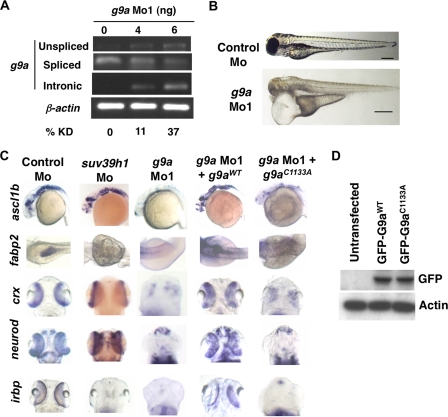
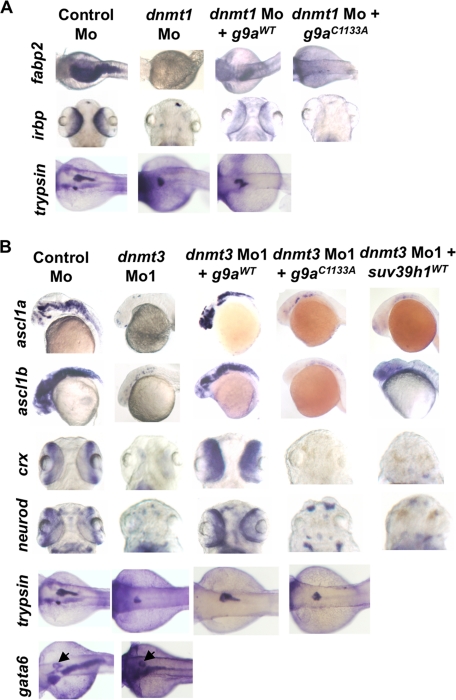
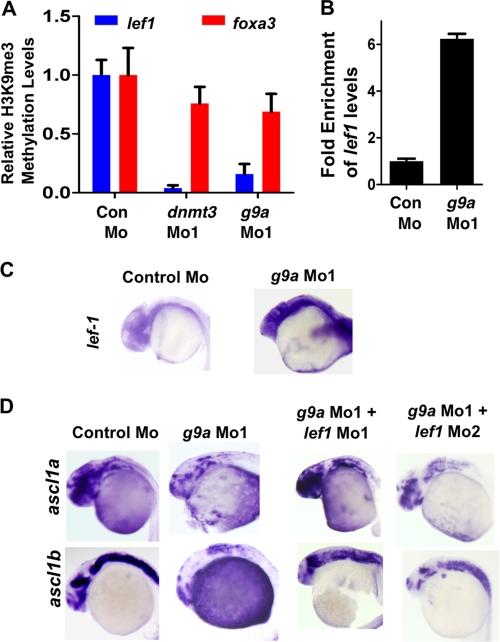
Although DNA methylation is critical for proper embryonic and tissue-specific development, how different DNA methyltransferases affect tissue-specific development and their targets remains unknown. We address this issue in zebrafish through antisense-based morpholino knockdown of Dnmt3 and Dnmt1. Our data reveal that Dnmt3 is required for proper neurogenesis, and its absence results in profound defects in brain and retina. Interestingly, other organs such as intestine remain unaffected suggesting tissue-specific requirements of Dnmt3. Further, comparison of Dnmt1 knockdown phenotypes with those of Dnmt3 suggested that these two families have distinct functions. Consistent with this idea, Dnmt1 failed to complement Dnmt3 deficiency, and Dnmt3 failed to complement Dnmt1 deficiency. Downstream of Dnmt3 we identify a neurogenesis regulator, lef1, as a Dnmt3-specific target gene that is demethylated and up-regulated in dnmt3 morphants. Knockdown of lef1 rescued neurogenesis defects resulting from Dnmt3 absence. Mechanistically, we show cooperation between Dnmt3 and an H3K9 methyltransferase G9a in regulating lef1. Further, like Dnmt1-Suv39h1 cooperativity, Dnmt3 and G9a seemed to function together for tissue-specific development. G9a knockdown, but not Suv39h1 loss, phenocopied dnmt3 morphants and G9a overexpression provided a striking rescue of dnmt3 morphant phenotypes, whereas Suv39h1 overexpression failed, supporting the notion of specific DNMT-histone methyltransferase networks. Consistent with this model, H3K9me3 levels on the lef1 promoter were reduced in both dnmt3 and g9a morphants, and its knockdown rescued neurogenesis defects in g9a morphants. We propose a model wherein specific DNMT-histone methyltransferase networks are utilized to silence critical regulators of cell fate in a tissue-specific manner.
虽然 DNA 甲基化对胚胎和组织特异性发育至关重要,但不同的 DNA 甲基转移酶如何影响组织特异性发育及其靶标尚不清楚。我们通过反义寡核苷酸对斑马鱼中的 Dnmt3 和 Dnmt1 进行基于形态的敲低来解决这个问题。我们的数据表明,Dnmt3 是神经发生所必需的,其缺失导致大脑和视网膜严重缺陷。有趣的是,其他器官如肠仍然不受影响,表明 Dnmt3 具有组织特异性需求。此外,Dnmt1 敲低表型与 Dnmt3 敲低表型的比较表明,这两个家族具有不同的功能。与这一观点一致的是,Dnmt1 未能补充 Dnmt3 的缺乏,而 Dnmt3 未能补充 Dnmt1 的缺乏。在 Dnmt3 下游,我们确定了一个神经发生调节剂 lef1 作为 Dnmt3 特异性靶基因,该基因在 dnmt3 形态发生缺陷体中被去甲基化和上调。 lef1 的敲低挽救了 Dnmt3 缺失引起的神经发生缺陷。从机制上讲,我们显示了 Dnmt3 与 H3K9 甲基转移酶 G9a 之间在调节 lef1 方面的合作。此外,与 Dnmt1-Suv39h1 合作一样,Dnmt3 和 G9a 似乎为组织特异性发育而共同发挥作用。G9a 敲低,但不是 Suv39h1 缺失,模拟了 dnmt3 形态发生缺陷体,而 G9a 过表达提供了 dnmt3 形态发生缺陷体表型的惊人挽救,而 Suv39h1 过表达则没有,支持了特定的 DNMT-组蛋白甲基转移酶网络的概念。与该模型一致,dnmt3 和 g9a 形态发生缺陷体中 lef1 启动子上的 H3K9me3 水平降低,其敲低挽救了 g9a 形态发生缺陷体中的神经发生缺陷。我们提出了一个模型,其中特定的 DNMT-组蛋白甲基转移酶网络被用于以组织特异性方式沉默细胞命运的关键调节剂。