Center for Cell Decision Processes, Boston, MA, USA.
Mol Syst Biol. 2009;5:331. doi: 10.1038/msb.2009.87. Epub 2009 Dec 1.
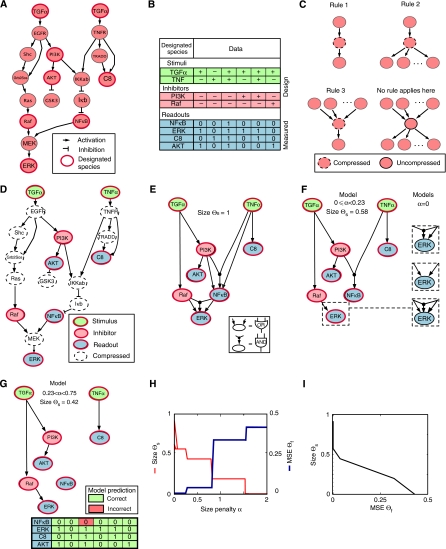
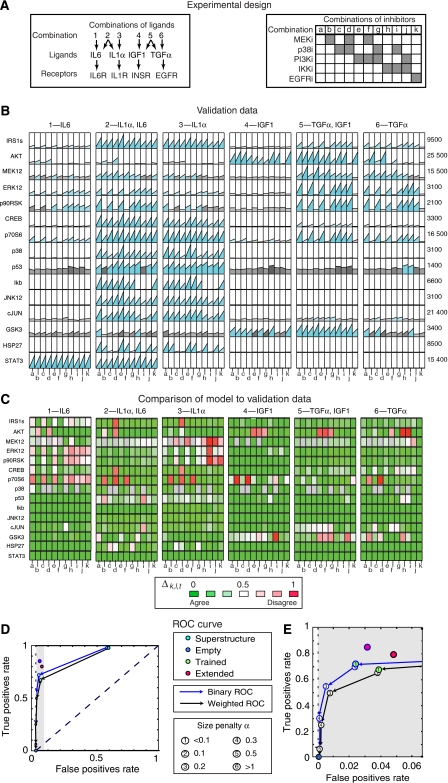
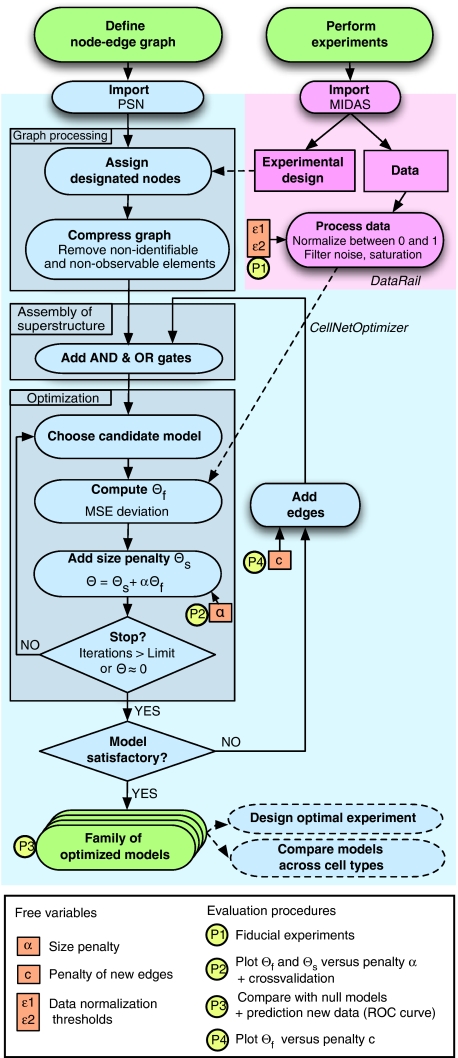
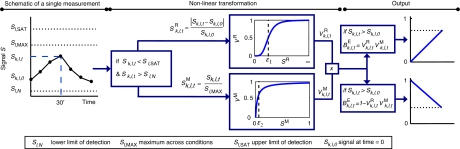
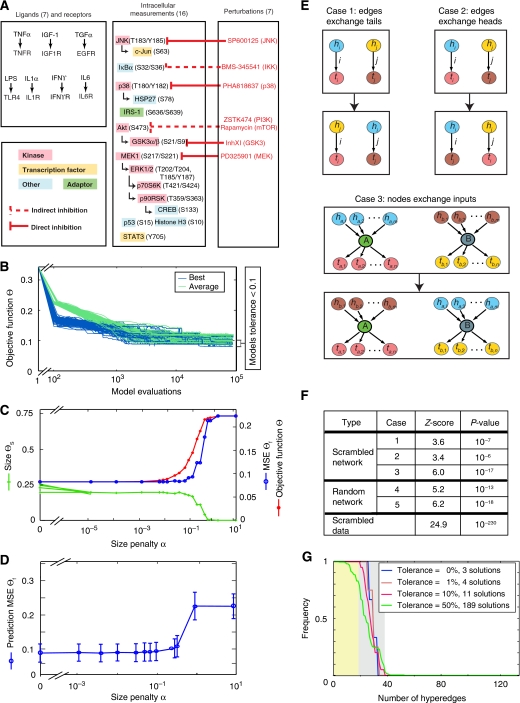
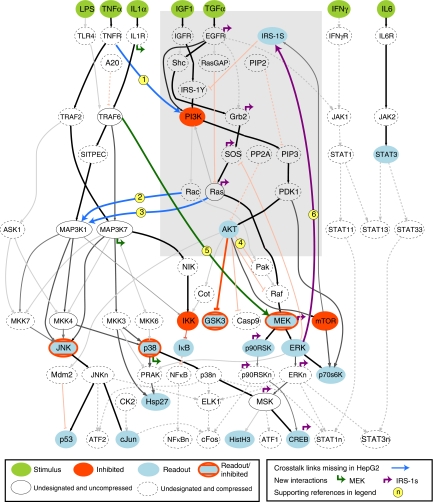
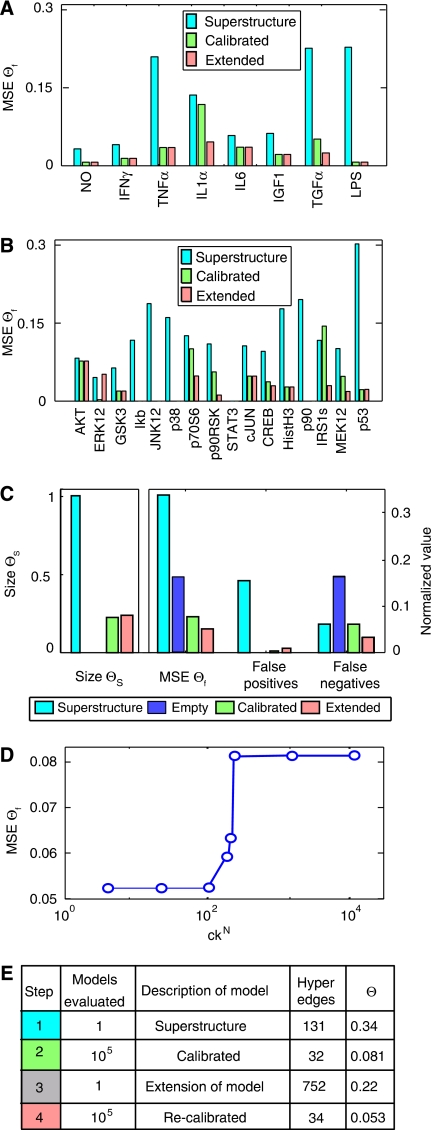
Large-scale protein signalling networks are useful for exploring complex biochemical pathways but do not reveal how pathways respond to specific stimuli. Such specificity is critical for understanding disease and designing drugs. Here we describe a computational approach--implemented in the free CNO software--for turning signalling networks into logical models and calibrating the models against experimental data. When a literature-derived network of 82 proteins covering the immediate-early responses of human cells to seven cytokines was modelled, we found that training against experimental data dramatically increased predictive power, despite the crudeness of Boolean approximations, while significantly reducing the number of interactions. Thus, many interactions in literature-derived networks do not appear to be functional in the liver cells from which we collected our data. At the same time, CNO identified several new interactions that improved the match of model to data. Although missing from the starting network, these interactions have literature support. Our approach, therefore, represents a means to generate predictive, cell-type-specific models of mammalian signalling from generic protein signalling networks.
大规模蛋白质信号网络可用于探索复杂的生化途径,但无法揭示途径如何对特定刺激做出响应。这种特异性对于理解疾病和设计药物至关重要。在这里,我们描述了一种计算方法——在免费的 CNO 软件中实现——将信号网络转化为逻辑模型,并根据实验数据对模型进行校准。当对涵盖人类细胞对七种细胞因子的即时早期反应的 82 种蛋白质的文献衍生网络进行建模时,我们发现尽管布尔近似值很粗糙,但通过针对实验数据进行训练,可极大地提高预测能力,同时还显著减少了相互作用的数量。因此,在我们收集数据的肝细胞中,文献衍生网络中的许多相互作用似乎没有功能。与此同时,CNO 确定了一些新的相互作用,可改善模型与数据的匹配度。尽管这些相互作用在起始网络中不存在,但它们具有文献支持。因此,我们的方法代表了一种从通用蛋白质信号网络生成预测性、细胞类型特异性哺乳动物信号模型的手段。