Biological Defense Research Directorate, Naval Medical Research Center, 503 Robert Grant Avenue, Silver Spring, Maryland 20910, USA.
Genome Biol. 2010 Jan 4;11(1):R1. doi: 10.1186/gb-2010-11-1-r1.
New DNA sequencing technologies have enabled detailed comparative genomic analyses of entire genera of bacterial pathogens. Prior to this study, three species of the enterobacterial genus Yersinia that cause invasive human diseases (Yersinia pestis, Yersinia pseudotuberculosis, and Yersinia enterocolitica) had been sequenced. However, there were no genomic data on the Yersinia species with more limited virulence potential, frequently found in soil and water environments.
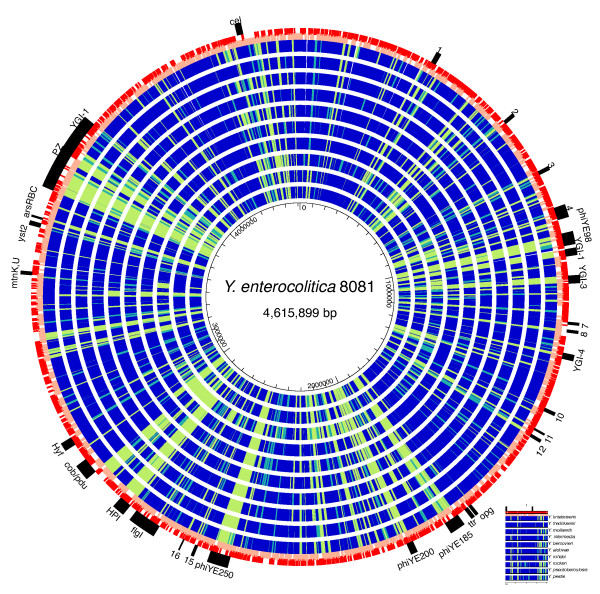
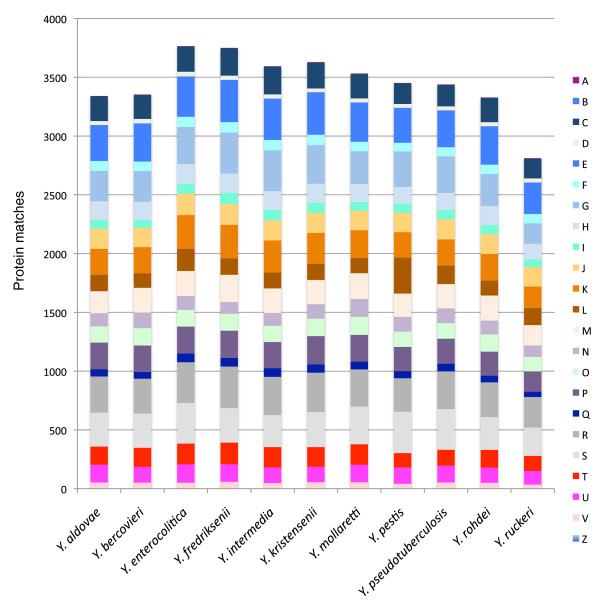
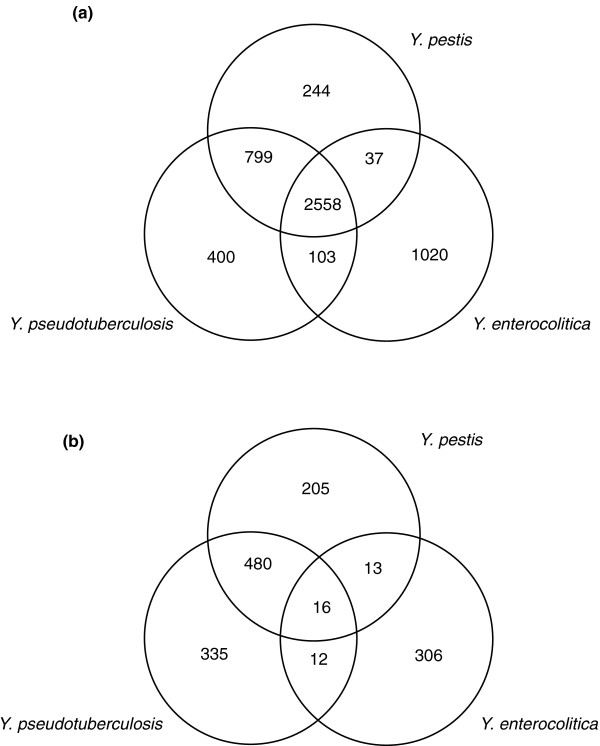
We used high-throughput sequencing-by-synthesis instruments to obtain 25- to 42-fold average redundancy, whole-genome shotgun data from the type strains of eight species: Y. aldovae, Y. bercovieri, Y. frederiksenii, Y. kristensenii, Y. intermedia, Y. mollaretii, Y. rohdei, and Y. ruckeri. The deepest branching species in the genus, Y. ruckeri, causative agent of red mouth disease in fish, has the smallest genome (3.7 Mb), although it shares the same core set of approximately 2,500 genes as the other members of the species, whose genomes range in size from 4.3 to 4.8 Mb. Yersinia genomes had a similar global partition of protein functions, as measured by the distribution of Cluster of Orthologous Groups families. Genome to genome variation in islands with genes encoding functions such as ureases, hydrogenases and B-12 cofactor metabolite reactions may reflect adaptations to colonizing specific host habitats.
Rapid high-quality draft sequencing was used successfully to compare pathogenic and non-pathogenic members of the Yersinia genus. This work underscores the importance of the acquisition of horizontally transferred genes in the evolution of Y. pestis and points to virulence determinants that have been gained and lost on multiple occasions in the history of the genus.
新的 DNA 测序技术使得对整个细菌病原体属的详细比较基因组分析成为可能。在这项研究之前,已经对引起侵袭性人类疾病的三种肠杆菌属耶尔森氏菌(鼠疫耶尔森氏菌、假结核耶尔森氏菌和小肠结肠炎耶尔森氏菌)进行了测序。然而,对于那些毒力潜力有限的耶尔森氏菌属,在土壤和水环境中经常发现,却没有基因组数据。
我们使用高通量测序合成仪器,从 8 个种的模式菌株中获得了 25 到 42 倍的平均冗余度、全基因组鸟枪法数据:Y. aldovae、Y. bercovieri、Y. frederiksenii、Y. kristensenii、Y. intermedia、Y. mollaretii、Y. rohdei 和 Y. ruckeri。在属中分支最深的物种 Y. ruckeri 是鱼类红嘴病的病原体,其基因组最小(3.7 Mb),尽管它与其他成员共享相同的大约 2500 个核心基因,其基因组大小范围从 4.3 到 4.8 Mb。耶尔森氏菌基因组具有相似的蛋白质功能全球分区,这可以通过同源基因簇家族的分布来衡量。编码脲酶、氢化酶和 B-12 辅因子代谢反应等功能的基因岛中的基因组到基因组的变异可能反映了对特定宿主栖息地的适应。
快速高质量的草图测序成功地用于比较致病性和非致病性的耶尔森氏菌属成员。这项工作强调了水平转移基因在鼠疫耶尔森氏菌进化中的重要性,并指出了在该属的历史上多次获得和失去的毒力决定因素。