Bioinformatics Core Facility, Swiss Institute of Bioinformatics, Génopode Building, Quartier Sorge, Lausanne CH-1015, Switzerland.
Breast Cancer Res. 2010;12(1):R5. doi: 10.1186/bcr2468. Epub 2010 Jan 11.
As part of the MicroArray Quality Control (MAQC)-II project, this analysis examines how the choice of univariate feature-selection methods and classification algorithms may influence the performance of genomic predictors under varying degrees of prediction difficulty represented by three clinically relevant endpoints.
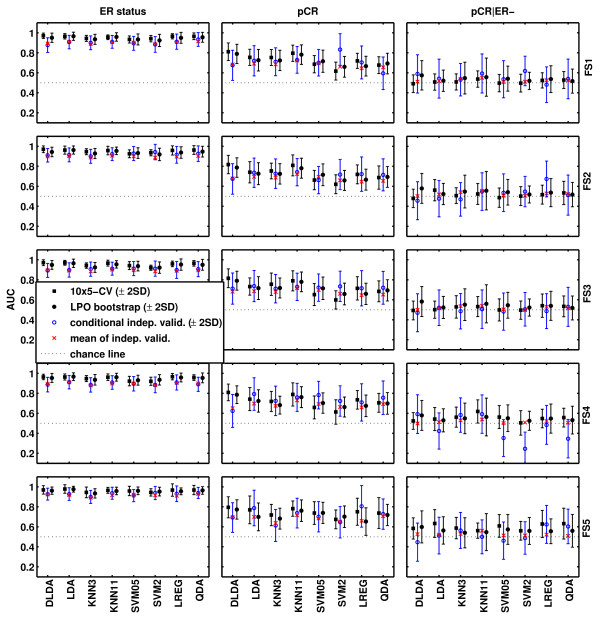
We used gene-expression data from 230 breast cancers (grouped into training and independent validation sets), and we examined 40 predictors (five univariate feature-selection methods combined with eight different classifiers) for each of the three endpoints. Their classification performance was estimated on the training set by using two different resampling methods and compared with the accuracy observed in the independent validation set.
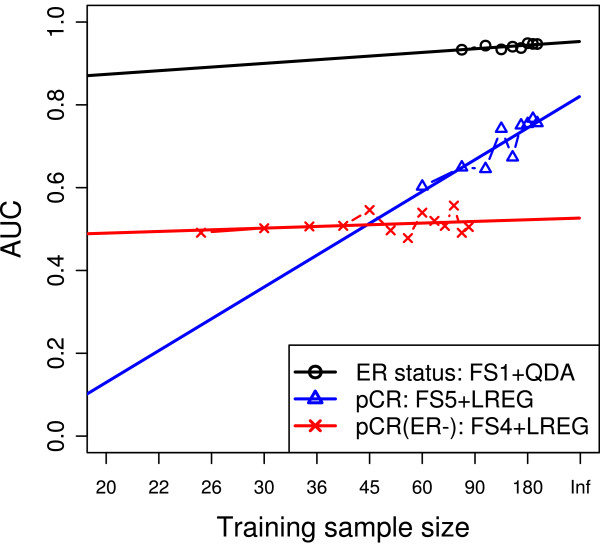
A ranking of the three classification problems was obtained, and the performance of 120 models was estimated and assessed on an independent validation set. The bootstrapping estimates were closer to the validation performance than were the cross-validation estimates. The required sample size for each endpoint was estimated, and both gene-level and pathway-level analyses were performed on the obtained models.
We showed that genomic predictor accuracy is determined largely by an interplay between sample size and classification difficulty. Variations on univariate feature-selection methods and choice of classification algorithm have only a modest impact on predictor performance, and several statistically equally good predictors can be developed for any given classification problem.
作为 MicroArray Quality Control(MAQC)-II 项目的一部分,本分析研究了在不同预测难度程度下(由三个临床相关终点表示),选择单变量特征选择方法和分类算法如何影响基因组预测器的性能。
我们使用了来自 230 个乳腺癌的基因表达数据(分为训练集和独立验证集),并针对每个三个终点,检查了 40 个预测器(五种单变量特征选择方法与八种不同的分类器结合)。使用两种不同的重采样方法在训练集上估计了它们的分类性能,并与独立验证集观察到的准确性进行了比较。
获得了三个分类问题的排名,并在独立验证集上估计和评估了 120 个模型的性能。与交叉验证估计相比,引导估计更接近验证性能。估计了每个终点所需的样本量,并对获得的模型进行了基因水平和途径水平的分析。
我们表明基因组预测器的准确性主要由样本量和分类难度之间的相互作用决定。单变量特征选择方法的变化和分类算法的选择对预测器性能的影响仅适度,对于任何给定的分类问题都可以开发出几个在统计学上同样好的预测器。