Center for Proteomics & Bioinformatics, Case Western Reserve University, Cleveland, Ohio, United States of America.
PLoS Comput Biol. 2010 Jan 15;6(1):e1000639. doi: 10.1371/journal.pcbi.1000639.
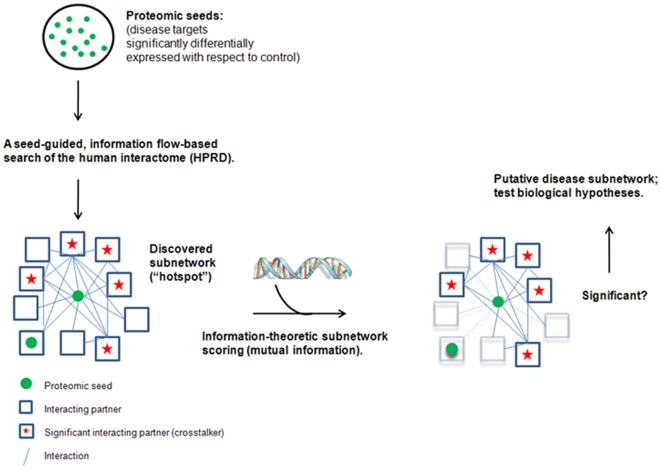
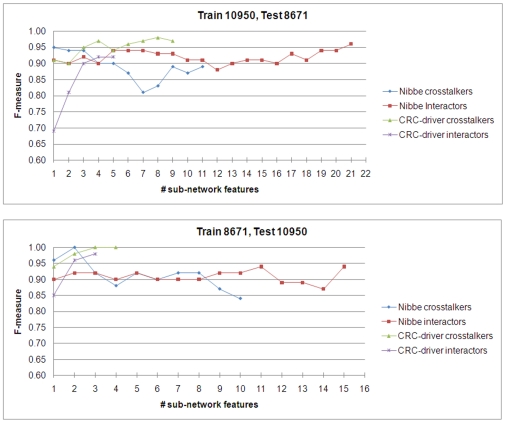
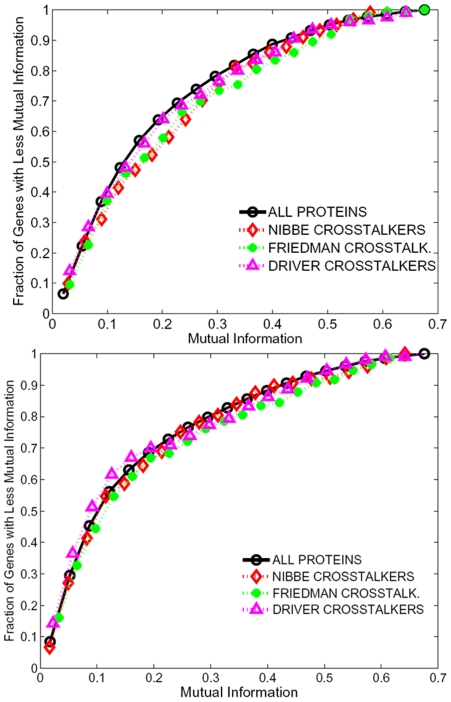
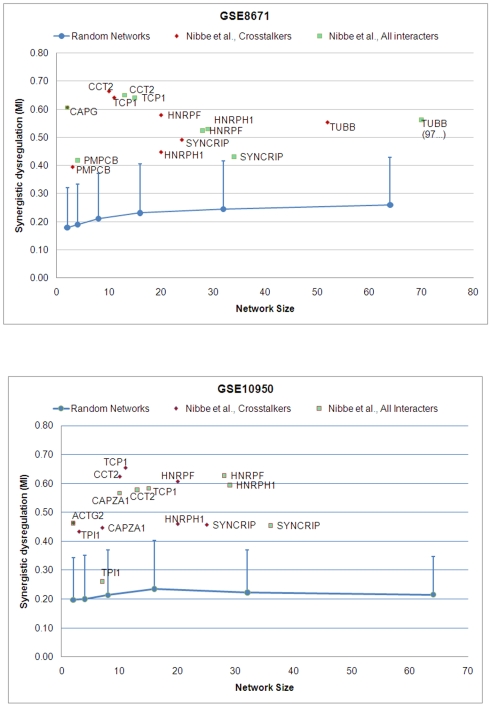
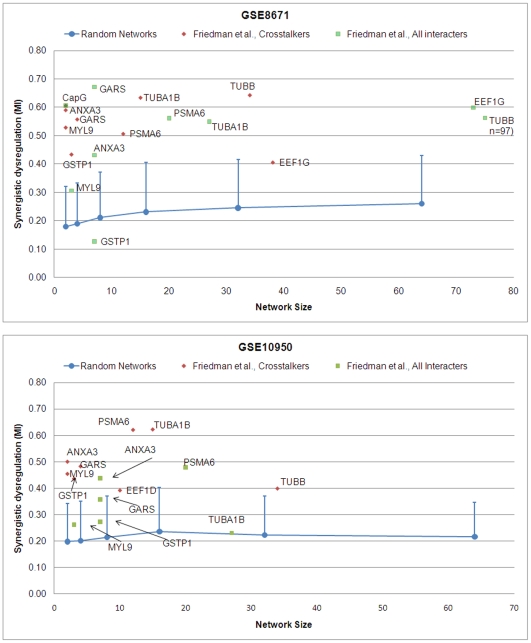
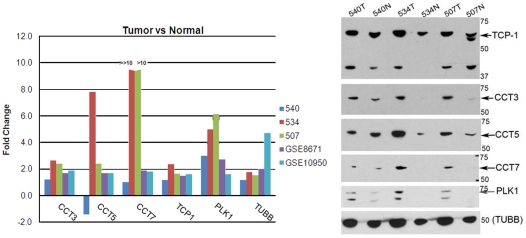
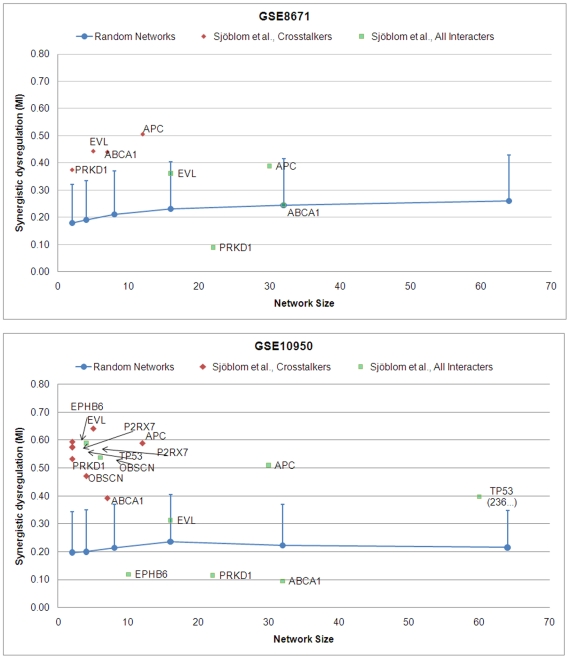
Emerging evidence indicates that gene products implicated in human cancers often cluster together in "hot spots" in protein-protein interaction (PPI) networks. Additionally, small sub-networks within PPI networks that demonstrate synergistic differential expression with respect to tumorigenic phenotypes were recently shown to be more accurate classifiers of disease progression when compared to single targets identified by traditional approaches. However, many of these studies rely exclusively on mRNA expression data, a useful but limited measure of cellular activity. Proteomic profiling experiments provide information at the post-translational level, yet they generally screen only a limited fraction of the proteome. Here, we demonstrate that integration of these complementary data sources with a "proteomics-first" approach can enhance the discovery of candidate sub-networks in cancer that are well-suited for mechanistic validation in disease. We propose that small changes in the mRNA expression of multiple genes in the neighborhood of a protein-hub can be synergistically associated with significant changes in the activity of that protein and its network neighbors. Further, we hypothesize that proteomic targets with significant fold change between phenotype and control may be used to "seed" a search for small PPI sub-networks that are functionally associated with these targets. To test this hypothesis, we select proteomic targets having significant expression changes in human colorectal cancer (CRC) from two independent 2-D gel-based screens. Then, we use random walk based models of network crosstalk and develop novel reference models to identify sub-networks that are statistically significant in terms of their functional association with these proteomic targets. Subsequently, using an information-theoretic measure, we evaluate synergistic changes in the activity of identified sub-networks based on genome-wide screens of mRNA expression in CRC. Cross-classification experiments to predict disease class show excellent performance using only a few sub-networks, underwriting the strength of the proposed approach in discovering relevant and reproducible sub-networks.
新兴证据表明,涉及人类癌症的基因产物经常聚集在蛋白质-蛋白质相互作用(PPI)网络的“热点”中。此外,最近还表明,PPI 网络中与肿瘤表型协同差异表达的小子网在预测疾病进展方面比传统方法确定的单个靶点更准确。然而,许多这些研究仅依赖于 mRNA 表达数据,这是一种有用但有限的细胞活动衡量标准。蛋白质组学分析实验提供了翻译后水平的信息,但它们通常仅筛选蛋白质组的有限部分。在这里,我们证明了将这些互补数据源与“以蛋白质组学为优先”的方法相结合,可以增强癌症候选子网的发现,这些子网非常适合在疾病中进行机制验证。我们提出,蛋白质枢纽附近多个基因的 mRNA 表达的微小变化可以与该蛋白质及其网络邻居的活性的显著变化协同相关。此外,我们假设蛋白质组学靶标在表型和对照之间具有显著的折叠变化,可用于“播种”搜索与这些靶标具有功能关联的小 PPI 子网。为了验证这一假设,我们从两个独立的二维凝胶基础筛选中选择了在人类结直肠癌(CRC)中具有显著表达变化的蛋白质组学靶标。然后,我们使用网络串扰的随机游走模型,并开发新的参考模型来识别在功能上与这些蛋白质组学靶标具有统计学意义的子网。随后,使用信息论度量,我们根据 CRC 中的全基因组 mRNA 表达筛选,评估所识别子网的活性的协同变化。仅使用少数子网进行交叉分类实验来预测疾病类别,表现出出色的性能,证明了所提出的方法在发现相关且可重复的子网方面的强大性。