Center for Bioinformatics ZBIT, Tübingen University, Sand 14, 72076 Tübingen, Germany.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2105-11-S1-S12.
Metagenomics is the study of environmental samples using sequencing. Rapid advances in sequencing technology are fueling a vast increase in the number and scope of metagenomics projects. Most metagenome sequencing projects so far have been based on Sanger or Roche-454 sequencing, as only these technologies provide long enough reads, while Illumina sequencing has not been considered suitable for metagenomic studies due to a short read length of only 35 bp. However, now that reads of length 75 bp can be sequenced in pairs, Illumina sequencing has become a viable option for metagenome studies.
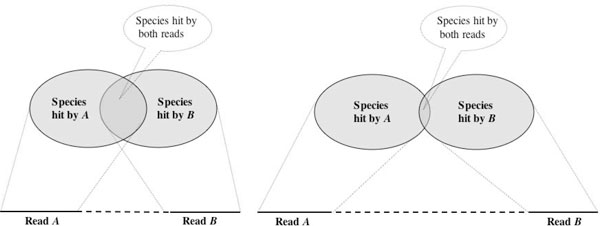
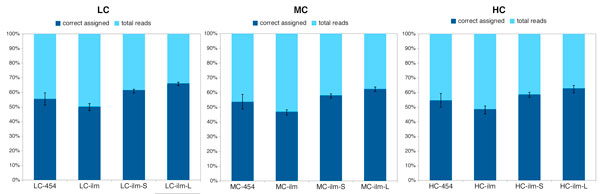
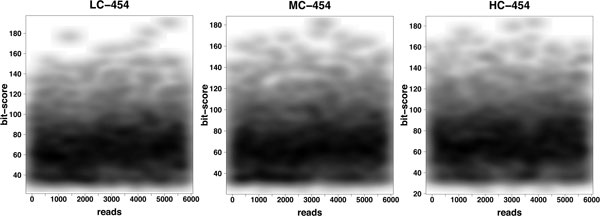
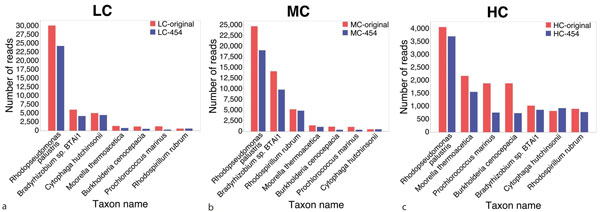
This paper addresses the problem of taxonomical analysis of paired reads. We describe a new feature of our metagenome analysis software MEGAN that allows one to process sequencing reads in pairs and makes assignments of such reads based on the combined bit scores of their matches to reference sequences. Using this new software in a simulation study, we investigate the use of Illumina paired-sequencing in taxonomical analysis and compare the performance of single reads, short clones and long clones. In addition, we also compare against simulated Roche-454 sequencing runs.
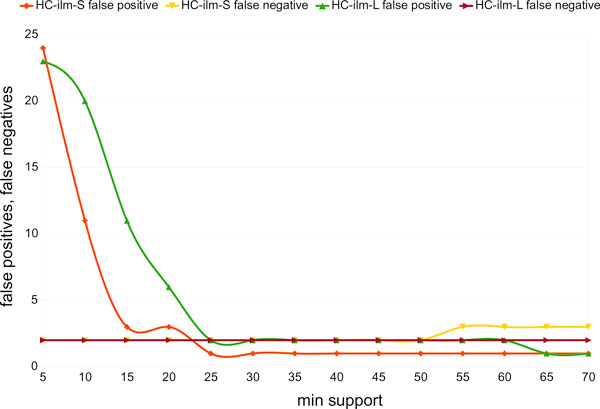
This work shows that paired reads perform better than single reads, as expected, but also, perhaps slightly less obviously, that long clones allow more specific assignments than short ones. A new version of the program MEGAN that explicitly takes paired reads into account is available from our website.
宏基因组学是使用测序技术研究环境样本的学科。测序技术的快速发展推动了宏基因组学项目数量和范围的大幅增加。到目前为止,大多数宏基因组测序项目都是基于 Sanger 或 Roche-454 测序的,因为只有这些技术能够提供足够长的读长,而 Illumina 测序由于读长仅为 35bp 而不适合宏基因组学研究。然而,现在可以对长度为 75bp 的读长进行双端测序,因此 Illumina 测序已经成为宏基因组学研究的一种可行选择。
本文解决了双端测序的分类分析问题。我们描述了我们的宏基因组分析软件 MEGAN 的一个新功能,该功能允许对测序读长进行双端处理,并基于其与参考序列匹配的综合位得分来对这些读长进行分配。使用这种新软件进行模拟研究,我们调查了 Illumina 双端测序在分类分析中的应用,并比较了单读长、短克隆和长克隆的性能。此外,我们还与模拟的 Roche-454 测序运行进行了比较。
这项工作表明,双端读长的表现优于单端读长,这是预期的结果,但也可能不太明显的是,长克隆比短克隆允许更具体的分配。我们的网站上提供了一个新版本的 MEGAN 程序,该程序明确考虑了双端读长。