Wu Shirley, Liu Tianyun, Altman Russ B
23andMe, 1390 Shorebird Way, Mountain View, CA, USA.
BMC Struct Biol. 2010 Feb 2;10:4. doi: 10.1186/1472-6807-10-4.
The emergence of structural genomics presents significant challenges in the annotation of biologically uncharacterized proteins. Unfortunately, our ability to analyze these proteins is restricted by the limited catalog of known molecular functions and their associated 3D motifs.
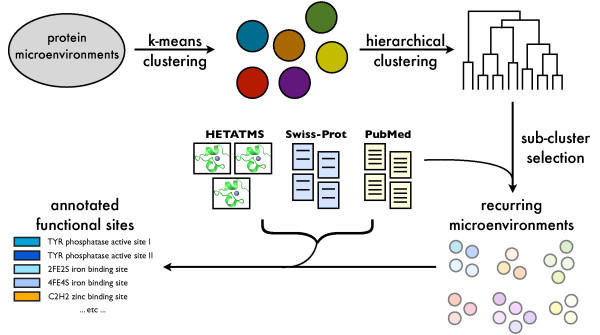
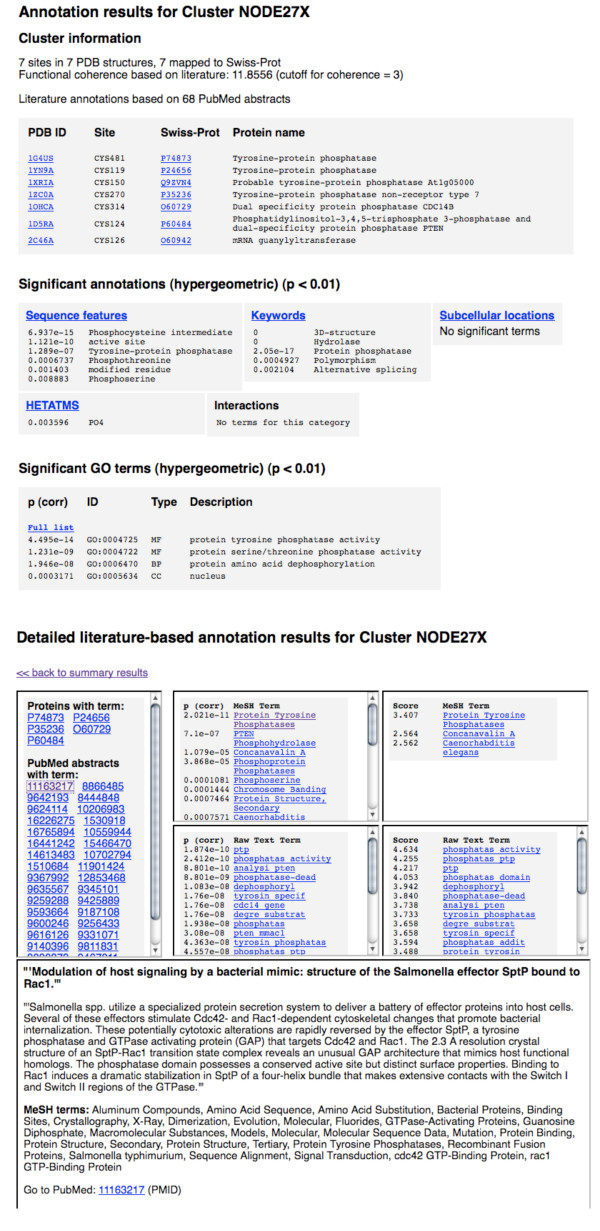
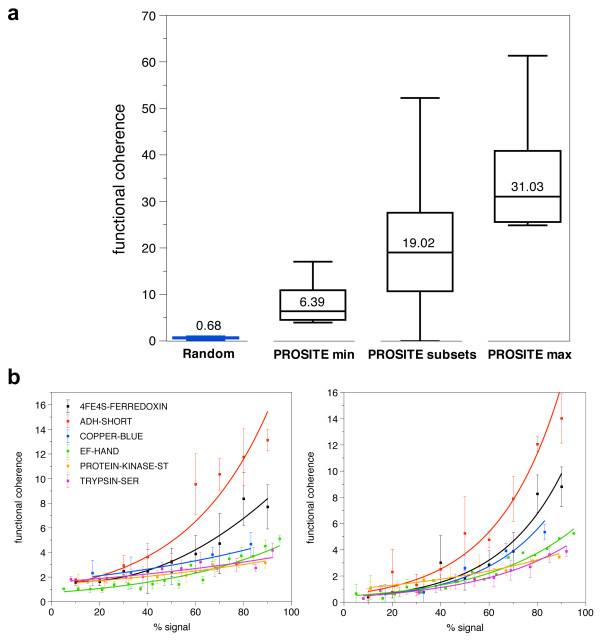
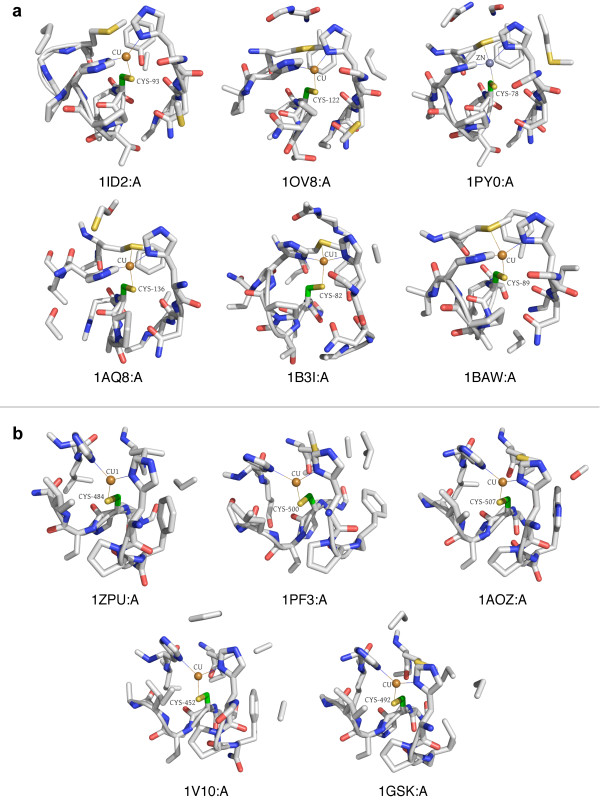
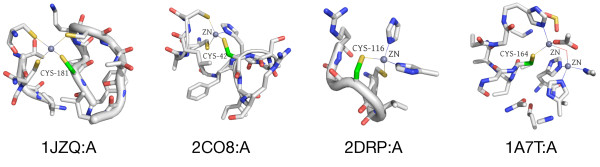
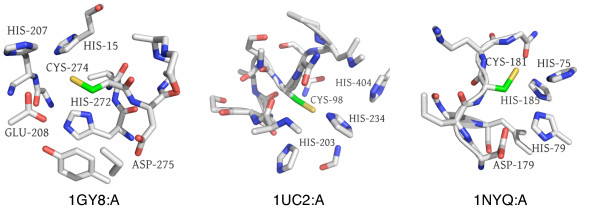
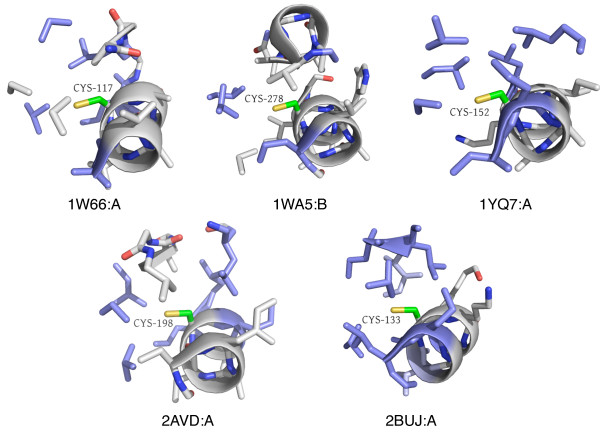
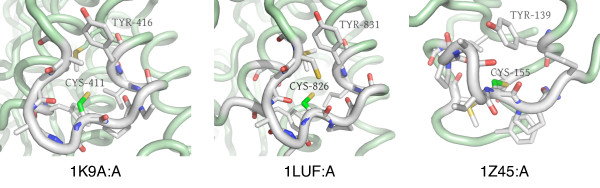
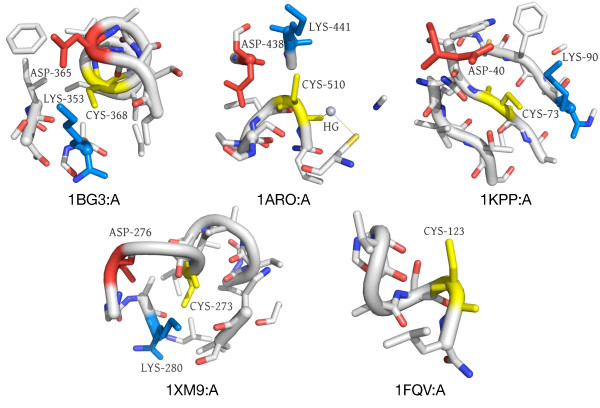
In order to identify novel 3D motifs that may be associated with molecular functions, we employ an unsupervised, two-phase clustering approach that combines k-means and hierarchical clustering with knowledge-informed cluster selection and annotation methods. We applied the approach to approximately 20,000 cysteine-based protein microenvironments (3D regions 7.5 A in radius) and identified 70 interesting clusters, some of which represent known motifs (e.g. metal binding and phosphatase activity), and some of which are novel, including several zinc binding sites. Detailed annotation results are available online for all 70 clusters at http://feature.stanford.edu/clustering/cys.
The use of microenvironments instead of backbone geometric criteria enables flexible exploration of protein function space, and detection of recurring motifs that are discontinuous in sequence and diverse in structure. Clustering microenvironments may thus help to functionally characterize novel proteins and better understand the protein structure-function relationship.
结构基因组学的出现给生物学特性未知的蛋白质注释带来了重大挑战。不幸的是,我们分析这些蛋白质的能力受到已知分子功能及其相关三维基序有限目录的限制。
为了识别可能与分子功能相关的新型三维基序,我们采用了一种无监督的两阶段聚类方法,该方法将k均值聚类和层次聚类与基于知识的聚类选择和注释方法相结合。我们将该方法应用于约20000个基于半胱氨酸的蛋白质微环境(半径为7.5埃的三维区域),并识别出70个有趣的聚类,其中一些代表已知基序(如金属结合和磷酸酶活性),一些是新型的,包括几个锌结合位点。所有70个聚类的详细注释结果可在http://feature.stanford.edu/clustering/cys在线获取。
使用微环境而非主链几何标准能够灵活地探索蛋白质功能空间,并检测序列中不连续且结构多样的重复基序。因此,对微环境进行聚类可能有助于对新型蛋白质进行功能表征,并更好地理解蛋白质结构与功能的关系。