Department of Genome Sciences, University of Washington, Box 355065, Seattle, Washington 98195, USA.
BMC Genomics. 2010 Feb 3;11:88. doi: 10.1186/1471-2164-11-88.
Experimental evolution of microbial populations provides a unique opportunity to study evolutionary adaptation in response to controlled selective pressures. However, until recently it has been difficult to identify the precise genetic changes underlying adaptation at a genome-wide scale. New DNA sequencing technologies now allow the genome of parental and evolved strains of microorganisms to be rapidly determined.
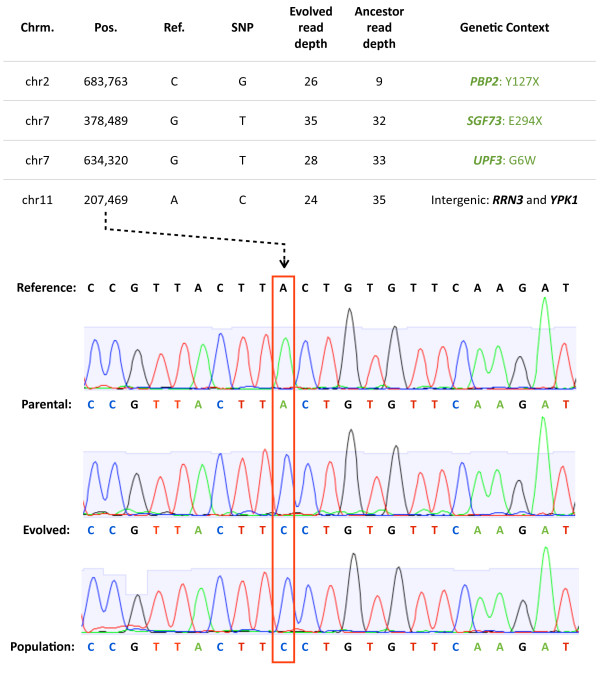
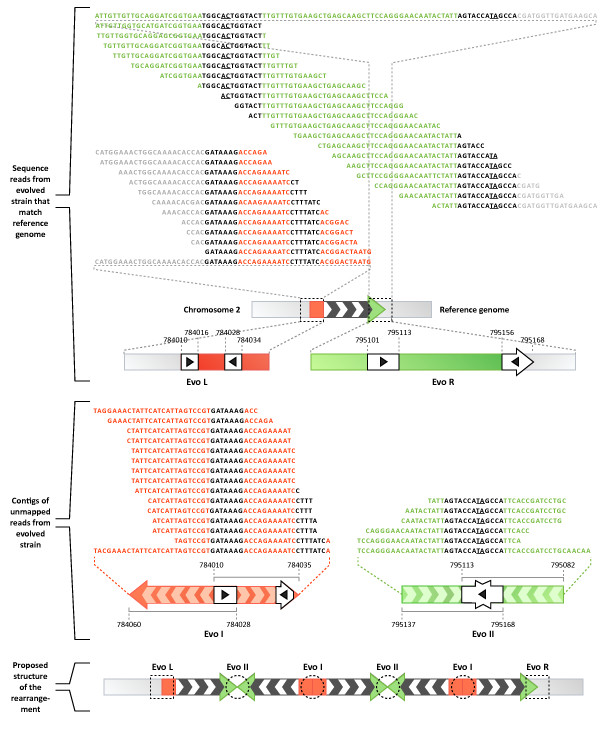
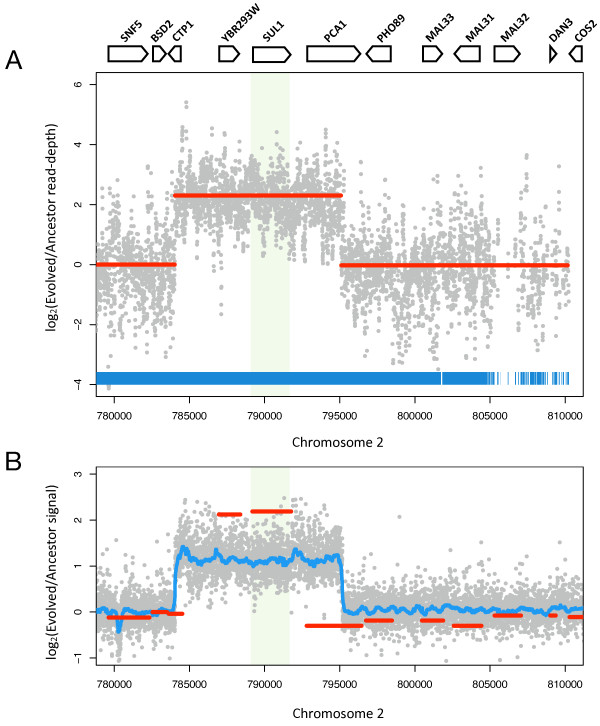
We sequenced >93.5% of the genome of a laboratory-evolved strain of the yeast Saccharomyces cerevisiae and its ancestor at >28x depth. Both single nucleotide polymorphisms and copy number amplifications were found, with specific gains over array-based methodologies previously used to analyze these genomes. Applying a segmentation algorithm to quantify structural changes, we determined the approximate genomic boundaries of a 5x gene amplification. These boundaries guided the recovery of breakpoint sequences, which provide insights into the nature of a complex genomic rearrangement.
This study suggests that whole-genome sequencing can provide a rapid approach to uncover the genetic basis of evolutionary adaptations, with further applications in the study of laboratory selections and mutagenesis screens. In addition, we show how single-end, short read sequencing data can provide detailed information about structural rearrangements, and generate predictions about the genomic features and processes that underlie genome plasticity.
微生物种群的实验进化为研究对受控选择压力的进化适应提供了独特的机会。然而,直到最近,要在全基因组范围内确定适应的精确遗传变化一直很困难。新的 DNA 测序技术现在可以快速确定微生物的亲本和进化菌株的基因组。
我们对实验室进化的酵母酿酒酵母及其祖先的菌株进行了测序,测序深度 >93.5%,达到 28 倍以上。我们发现了单核苷酸多态性和拷贝数扩增,与以前用于分析这些基因组的基于阵列的方法相比,有特定的增益。应用分割算法来量化结构变化,我们确定了 5x 基因扩增的大致基因组边界。这些边界指导了断点序列的恢复,为复杂基因组重排的性质提供了深入的见解。
本研究表明,全基因组测序可以快速揭示进化适应的遗传基础,在实验室选择和诱变筛选的研究中有进一步的应用。此外,我们展示了如何使用单端短读测序数据提供有关结构重排的详细信息,并对基因组可塑性的基础基因组特征和过程进行预测。