Pandey Ram Vinay, Nolte Viola, Schlötterer Christian
Institut für Populationsgenetik, Veterinärmedizinische Universität Wien, Veterinärplatz 1, Vienna, Austria.
BMC Res Notes. 2010 Jan 11;3:3. doi: 10.1186/1756-0500-3-3.
Next generation sequencing (NGS) technologies have substantially increased the sequence output while the costs were dramatically reduced. In addition to the use in whole genome sequencing, the 454 GS-FLX platform is becoming a widely used tool for biodiversity surveys based on amplicon sequencing. In order to use NGS for biodiversity surveys, software tools are required, which perform quality control, trimming of the sequence reads, removal of PCR primers, and generation of input files for downstream analyses. A user-friendly software utility that carries out these steps is still lacking.
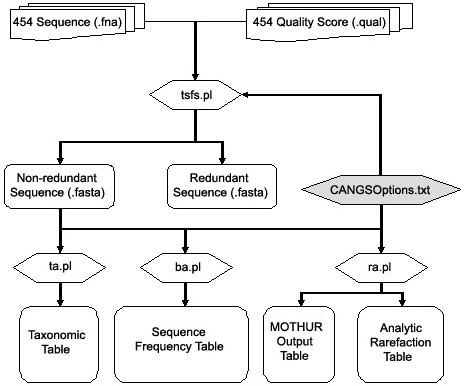
We developed CANGS (Cleaning and Analyzing Next Generation Sequences) a flexible and user-friendly integrated software utility: CANGS is designed for amplicon based biodiversity surveys using the 454 sequencing platform. CANGS filters low quality sequences, removes PCR primers, filters singletons, identifies barcodes, and generates input files for downstream analyses. The downstream analyses rely either on third party software (e.g.: rarefaction analyses) or CANGS-specific scripts. The latter include modules linking 454 sequences with the name of the closest taxonomic reference retrieved from the NCBI database and the sequence divergence between them. Our software can be easily adapted to handle sequencing projects with different amplicon sizes, primer sequences, and quality thresholds, which makes this software especially useful for non-bioinformaticians.
CANGS performs PCR primer clipping, filtering of low quality sequences, links sequences to NCBI taxonomy and provides input files for common rarefaction analysis software programs. CANGS is written in Perl and runs on Mac OS X/Linux and is available at http://i122server.vu-wien.ac.at/pop/software.html.
新一代测序(NGS)技术在大幅增加测序产出的同时,成本也显著降低。除了用于全基因组测序外,454 GS - FLX平台正成为基于扩增子测序进行生物多样性调查的广泛使用工具。为了将NGS用于生物多样性调查,需要软件工具来进行质量控制、修剪序列读数、去除PCR引物以及生成用于下游分析的输入文件。目前仍缺乏一个执行这些步骤的用户友好型软件实用程序。
我们开发了CANGS(下一代序列清理与分析),这是一个灵活且用户友好的集成软件实用程序:CANGS专为使用454测序平台基于扩增子的生物多样性调查而设计。CANGS可过滤低质量序列、去除PCR引物、过滤单例序列、识别条形码,并生成用于下游分析的输入文件。下游分析可依赖第三方软件(如:稀疏化分析)或CANGS特定脚本。后者包括将454序列与从NCBI数据库检索到的最接近分类学参考名称及其之间的序列差异相链接的模块。我们的软件可以轻松适应处理具有不同扩增子大小、引物序列和质量阈值的测序项目,这使得该软件对非生物信息学家特别有用。
CANGS执行PCR引物剪切、低质量序列过滤、将序列与NCBI分类学相链接,并为常见的稀疏化分析软件程序提供输入文件。CANGS用Perl编写,可在Mac OS X/Linux上运行,可从http://i122server.vu - wien.ac.at/pop/software.html获取。