Department of Veterinary Science, The University of Melbourne, Werribee, Victoria, Australia.
PLoS Negl Trop Dis. 2010 May 11;4(5):e684. doi: 10.1371/journal.pntd.0000684.
The blood-feeding hookworm Necator americanus infects hundreds of millions of people worldwide. In order to elucidate fundamental molecular biological aspects of this hookworm, the transcriptome of the adult stage of Necator americanus was explored using next-generation sequencing and bioinformatic analyses.
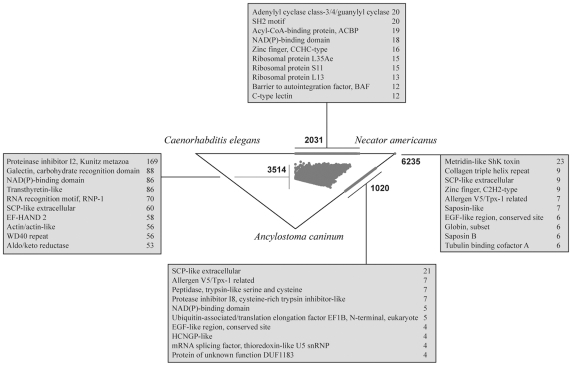
METHODOLOGY/PRINCIPAL FINDINGS: A total of 19,997 contigs were assembled from the sequence data; 6,771 of these contigs had known orthologues in the free-living nematode Caenorhabditis elegans, and most of them encoded proteins with WD40 repeats (10.6%), proteinase inhibitors (7.8%) or calcium-binding EF-hand proteins (6.7%). Bioinformatic analyses inferred that the C. elegans homologues are involved mainly in biological pathways linked to ribosome biogenesis (70%), oxidative phosphorylation (63%) and/or proteases (60%); most of these molecules were predicted to be involved in more than one biological pathway. Comparative analyses of the transcriptomes of N. americanus and the canine hookworm, Ancylostoma caninum, revealed qualitative and quantitative differences. For instance, proteinase inhibitors were inferred to be highly represented in the former species, whereas SCP/Tpx-1/Ag5/PR-1/Sc7 proteins ( = SCP/TAPS or Ancylostoma-secreted proteins) were predominant in the latter. In N. americanus, essential molecules were predicted using a combination of orthology mapping and functional data available for C. elegans. Further analyses allowed the prioritization of 18 predicted drug targets which did not have homologues in the human host. These candidate targets were inferred to be linked to mitochondrial (e.g., processing proteins) or amino acid metabolism (e.g., asparagine t-RNA synthetase).
This study has provided detailed insights into the transcriptome of the adult stage of N. americanus and examines similarities and differences between this species and A. caninum. Future efforts should focus on comparative transcriptomic and proteomic investigations of the other predominant human hookworm, A. duodenale, for both fundamental and applied purposes, including the prevalidation of anti-hookworm drug targets.
寄生人体的钩虫——美洲板口线虫感染了全球数亿人。为了阐明这种钩虫的基本分子生物学特性,我们利用下一代测序和生物信息学分析方法,对美洲板口线虫成虫的转录组进行了研究。
方法/主要发现:从序列数据中组装了 19997 个连续序列;其中 6771 个连续序列在自由生活的线虫秀丽隐杆线虫中有已知的直系同源物,它们大多数编码含有 WD40 重复(10.6%)、蛋白酶抑制剂(7.8%)或钙结合 EF 手蛋白(6.7%)的蛋白。生物信息学分析推断,秀丽隐杆线虫的同源物主要参与核糖体生物发生(70%)、氧化磷酸化(63%)和/或蛋白酶(60%)相关的生物途径;这些分子大多数被预测参与不止一个生物途径。对美洲板口线虫和犬钩虫的转录组进行比较分析,揭示了它们之间存在定性和定量的差异。例如,蛋白酶抑制剂被推断在前者中高度表达,而 SCP/Tpx-1/Ag5/PR-1/Sc7 蛋白(= SCP/TAPS 或Ancylostoma-secreted proteins)在后者中占主导地位。在美洲板口线虫中,利用与秀丽隐杆线虫的同源映射和功能数据相结合的方法,预测了必需分子。进一步的分析允许对 18 个没有在人类宿主中发现同源物的预测药物靶标进行优先级排序。这些候选靶标被推断与线粒体(例如,加工蛋白)或氨基酸代谢(例如,天冬酰胺 t-RNA 合成酶)有关。
本研究详细阐述了美洲板口线虫成虫的转录组,并考察了该物种与犬钩虫之间的相似性和差异性。未来的研究应集中于对其他主要的人体钩虫——十二指肠钩虫的比较转录组和蛋白质组学研究,这不仅对基础研究具有重要意义,也对应用研究具有重要意义,包括抗钩虫药物靶标的预验证。