Shi Zhiao, Derow Catherine K, Zhang Bing
Advanced Computing Center for Research & Education, Vanderbilt University, Nashville, TN 37240, USA.
BMC Syst Biol. 2010 May 27;4:74. doi: 10.1186/1752-0509-4-74.
Gene expression signatures are typically identified by correlating gene expression patterns to a disease phenotype of interest. However, individual gene-based signatures usually suffer from low reproducibility and interpretability.
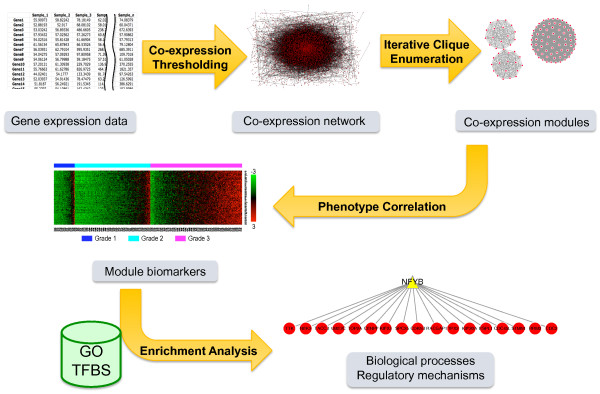
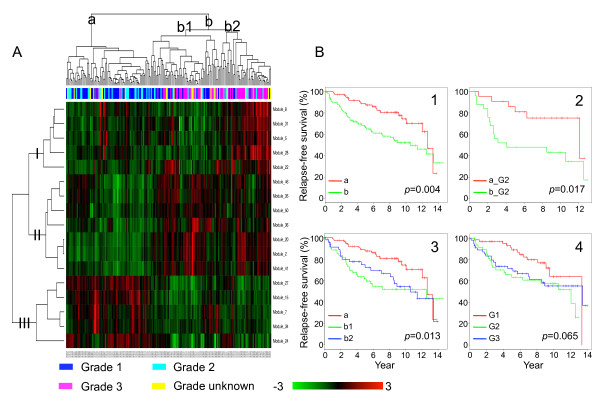
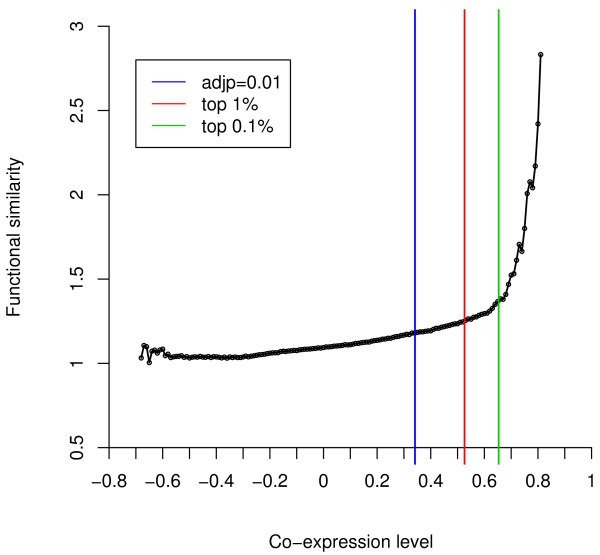
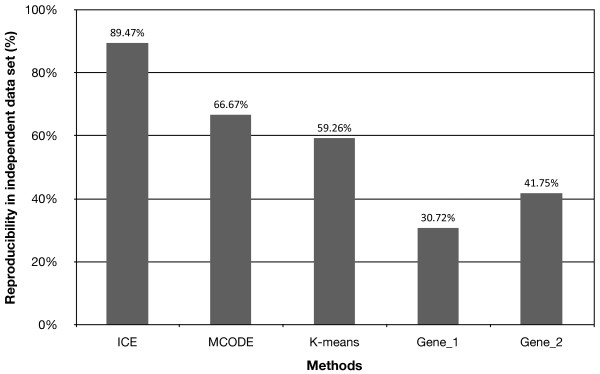
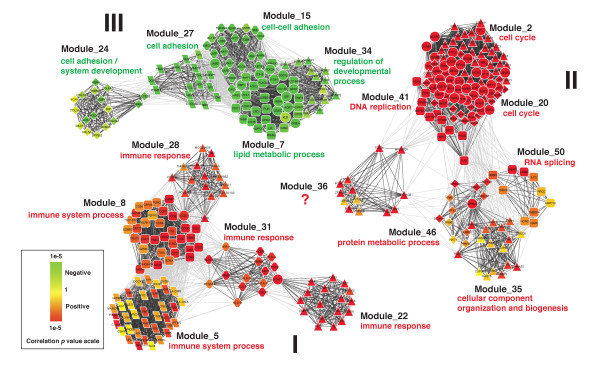
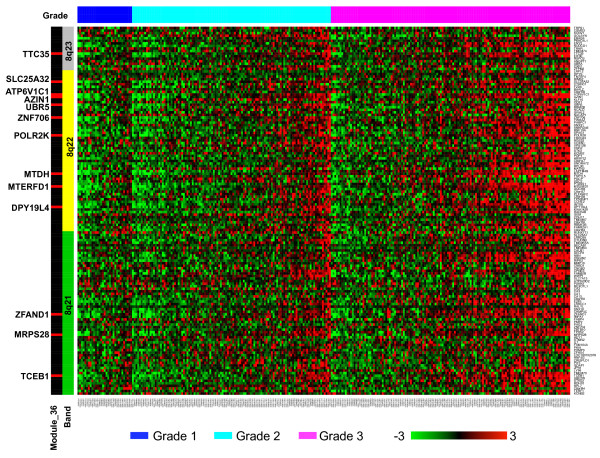
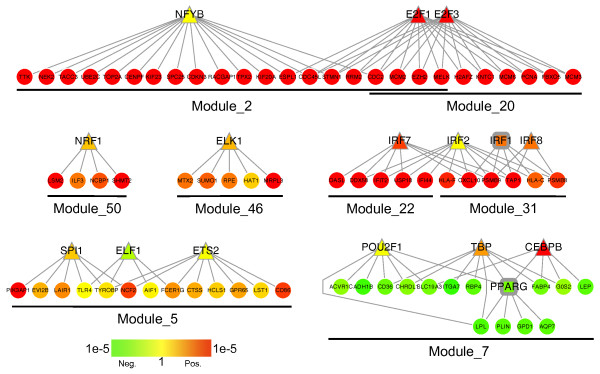
We have developed a novel algorithm Iterative Clique Enumeration (ICE) for identifying relatively independent maximal cliques as co-expression modules and a module-based approach to the analysis of gene expression data. Applying this approach on a public breast cancer dataset identified 19 modules whose expression levels were significantly correlated with tumor grade. The correlations were reproducible for 17 modules in an independent breast cancer dataset, and the reproducibility was considerably higher than that based on individual genes or modules identified by other algorithms. Sixteen out of the 17 modules showed significant enrichment in certain Gene Ontology (GO) categories. Specifically, modules related to cell proliferation and immune response were up-regulated in high-grade tumors while those related to cell adhesion was down-regulated. Further analyses showed that transcription factors NYFB, E2F1/E2F3, NRF1, and ELK1 were responsible for the up-regulation of the cell proliferation modules. IRF family and ETS family proteins were responsible for the up-regulation of the immune response modules. Moreover, inhibition of the PPARA signaling pathway may also play an important role in tumor progression. The module without GO enrichment was found to be associated with a potential genomic gain in 8q21-23 in high-grade tumors. The 17-module signature of breast tumor progression clustered patients into subgroups with significantly different relapse-free survival times. Namely, patients with lower cell proliferation and higher cell adhesion levels had significantly lower risk of recurrence, both for all patients (p = 0.004) and for those with grade 2 tumors (p = 0.017).
The ICE algorithm is effective in identifying relatively independent co-expression modules from gene co-expression networks and the module-based approach illustrated in this study provides a robust, interpretable, and mechanistic characterization of transcriptional changes.
基因表达特征通常通过将基因表达模式与感兴趣的疾病表型相关联来识别。然而,基于单个基因的特征通常具有低重现性和低可解释性。
我们开发了一种名为迭代团枚举(ICE)的新算法,用于识别相对独立的最大团作为共表达模块,并开发了一种基于模块的方法来分析基因表达数据。将此方法应用于一个公开的乳腺癌数据集,识别出19个模块,其表达水平与肿瘤分级显著相关。在一个独立的乳腺癌数据集中,17个模块的相关性具有可重复性,且该重现性显著高于基于其他算法识别的单个基因或模块的重现性。17个模块中有16个在某些基因本体(GO)类别中显示出显著富集。具体而言,与细胞增殖和免疫反应相关的模块在高级别肿瘤中上调,而与细胞黏附相关的模块下调。进一步分析表明,转录因子NYFB、E2F1/E2F3、NRF1和ELK1负责细胞增殖模块的上调。IRF家族和ETS家族蛋白负责免疫反应模块的上调。此外,PPARA信号通路的抑制在肿瘤进展中可能也起重要作用。发现无GO富集的模块与高级别肿瘤中8q21 - 23区域潜在的基因组增益相关。乳腺癌进展的17模块特征将患者聚类为无复发生存时间显著不同的亚组。即,对于所有患者(p = 0.004)以及2级肿瘤患者(p = 0.017),细胞增殖水平较低且细胞黏附水平较高的患者复发风险显著较低。
ICE算法可有效地从基因共表达网络中识别相对独立的共表达模块,本研究中展示的基于模块的方法为转录变化提供了稳健、可解释且具机制性的特征描述。