Department of Plant Systems Biology, VIB, Ghent, Belgium.
BMC Genomics. 2010 Jun 3;11:353. doi: 10.1186/1471-2164-11-353.
Oomycetes of the genus Phytophthora are pathogens that infect a wide range of plant species. For dicot hosts such as tomato, potato and soybean, Phytophthora is even the most important pathogen. Previous analyses of Phytophthora genomes uncovered many genes, large gene families and large genome sizes that can partially be explained by significant repeat expansion patterns.
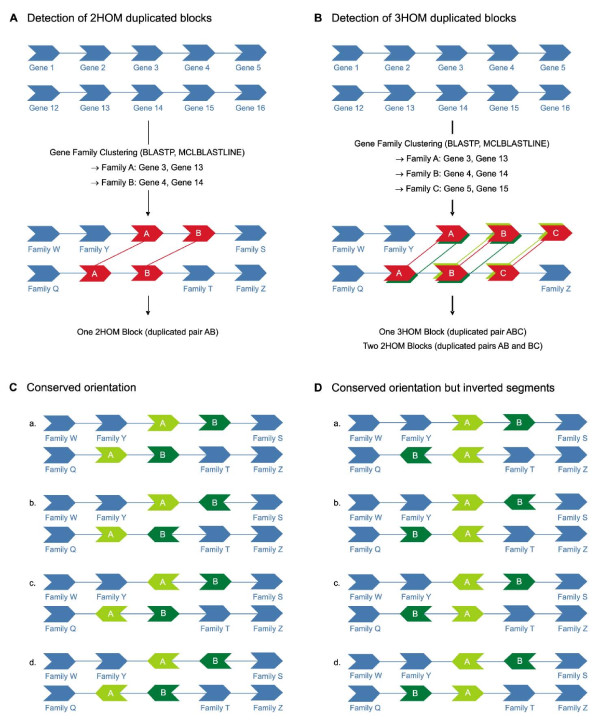
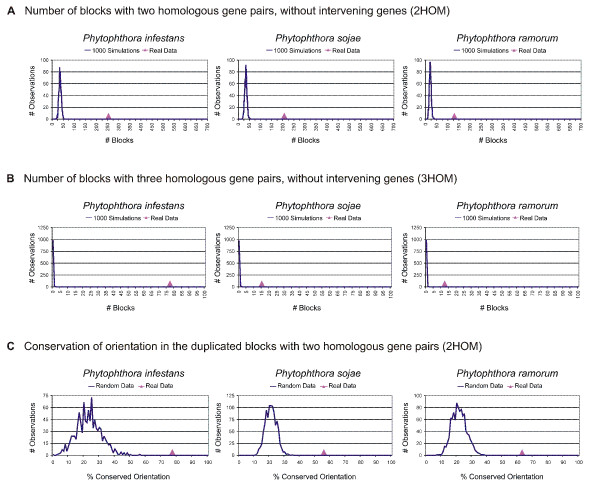
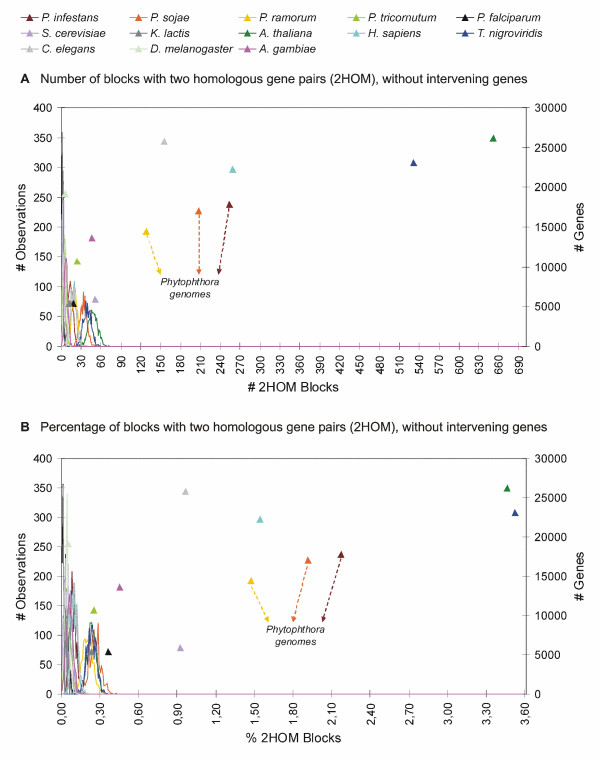
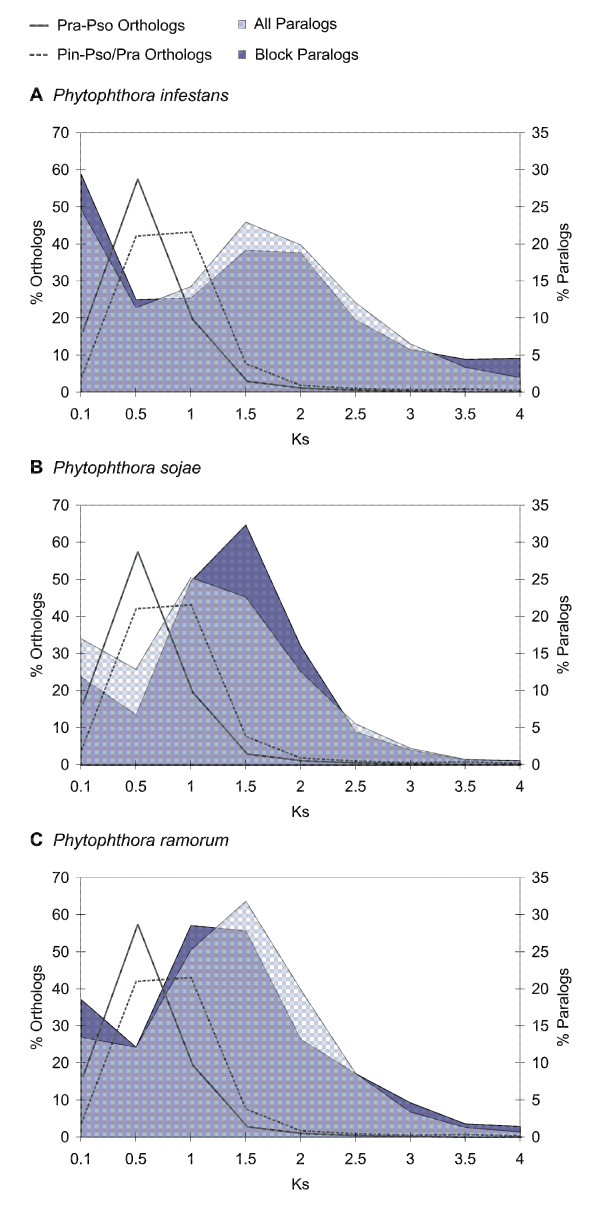
Analysis of the complete genomes of three different Phytophthora species, using a newly developed approach, unveiled a large number of small duplicated blocks, mainly consisting of two or three consecutive genes. Further analysis of these duplicated genes and comparison with the known gene and genome duplication history of ten other eukaryotes including parasites, algae, plants, fungi, vertebrates and invertebrates, suggests that the ancestor of P. infestans, P. sojae and P. ramorum most likely underwent a whole genome duplication (WGD). Genes that have survived in duplicate are mainly genes that are known to be preferentially retained following WGDs, but also genes important for pathogenicity and infection of the different hosts seem to have been retained in excess. As a result, the WGD might have contributed to the evolutionary and pathogenic success of Phytophthora.
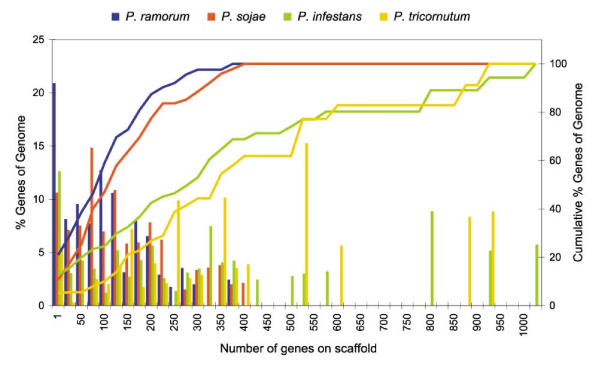
The fact that we find many small blocks of duplicated genes indicates that the genomes of Phytophthora species have been heavily rearranged following the WGD. Most likely, the high repeat content in these genomes have played an important role in this rearrangement process. As a consequence, the paucity of retained larger duplicated blocks has greatly complicated previous attempts to detect remnants of a large-scale duplication event in Phytophthora. However, as we show here, our newly developed strategy to identify very small duplicated blocks might be a useful approach to uncover ancient polyploidy events, in particular for heavily rearranged genomes.
疫霉属的卵菌是一类感染范围广泛的植物物种的病原体。对于双子叶宿主,如番茄、土豆和大豆,卵菌甚至是最重要的病原体。以前对卵菌基因组的分析揭示了许多基因、大基因家族和大基因组大小,这些可以部分解释为显著的重复扩张模式。
使用新开发的方法分析了三个不同的卵菌物种的完整基因组,揭示了大量的小重复块,主要由两个或三个连续的基因组成。对这些重复基因的进一步分析,并与包括寄生虫、藻类、植物、真菌、脊椎动物和无脊椎动物在内的其他十个真核生物的已知基因和基因组重复历史进行比较,表明晚疫病菌、大豆疫霉和松材线虫的祖先很可能经历了全基因组复制(WGD)。在重复中幸存下来的基因主要是已知在 WGD 后优先保留的基因,但对不同宿主的致病性和感染也很重要的基因似乎也被过度保留了。因此,WGD 可能有助于卵菌的进化和致病性成功。
我们发现许多小的重复基因块的事实表明,卵菌物种的基因组在 WGD 后经历了严重的重排。很可能,这些基因组中的高重复含量在这个重排过程中发挥了重要作用。因此,保留的较大重复块的缺乏极大地复杂化了以前在卵菌中检测到大规模复制事件痕迹的尝试。然而,正如我们在这里所展示的,我们新开发的识别非常小的重复块的策略可能是揭示古老多倍体事件的有用方法,特别是对于重排的基因组。