Department of Computer Science, San Diego State University, CA, USA.
BMC Bioinformatics. 2010 Jun 23;11:341. doi: 10.1186/1471-2105-11-341.
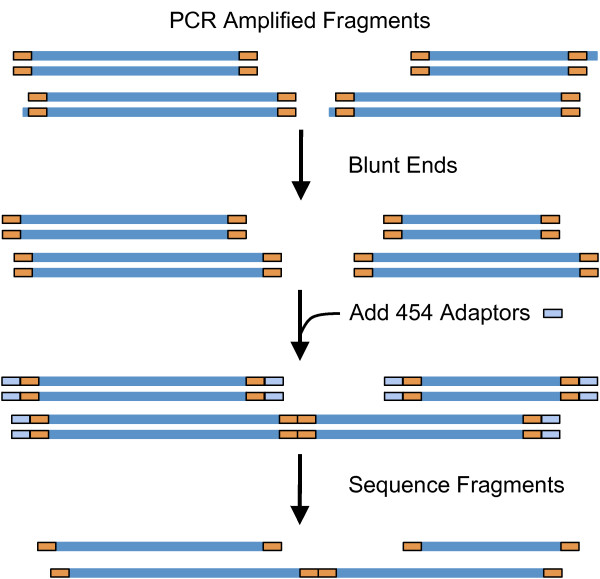
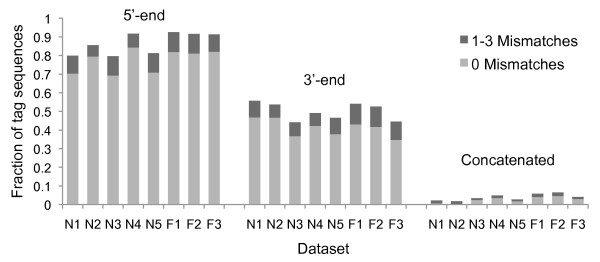
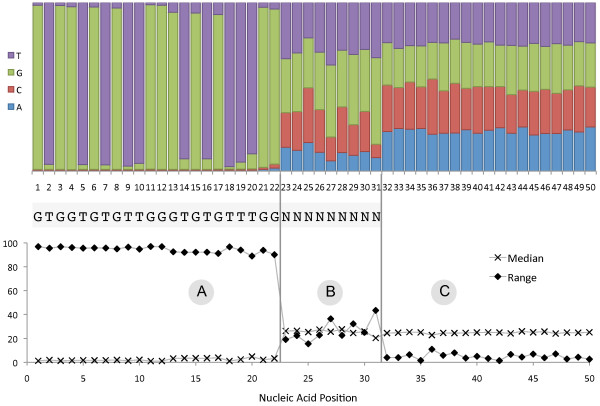
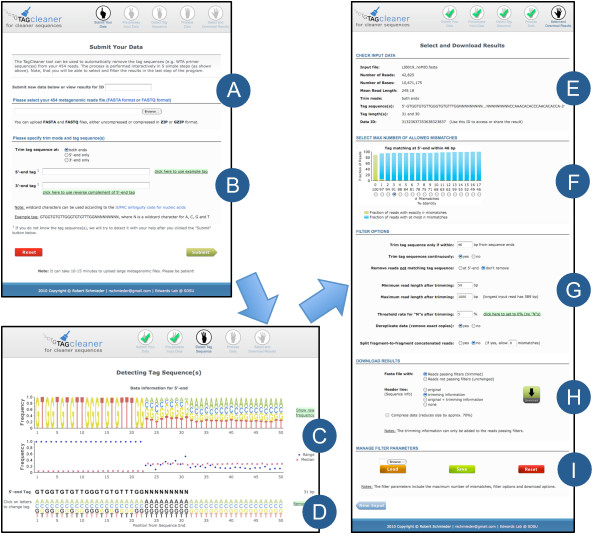
Sequencing metagenomes that were pre-amplified with primer-based methods requires the removal of the additional tag sequences from the datasets. The sequenced reads can contain deletions or insertions due to sequencing limitations, and the primer sequence may contain ambiguous bases. Furthermore, the tag sequence may be unavailable or incorrectly reported. Because of the potential for downstream inaccuracies introduced by unwanted sequence contaminations, it is important to use reliable tools for pre-processing sequence data.
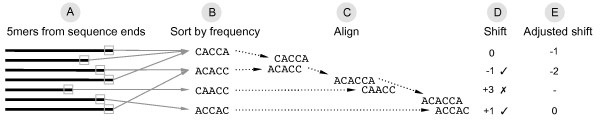
TagCleaner is a web application developed to automatically identify and remove known or unknown tag sequences allowing insertions and deletions in the dataset. TagCleaner is designed to filter the trimmed reads for duplicates, short reads, and reads with high rates of ambiguous sequences. An additional screening for and splitting of fragment-to-fragment concatenations that gave rise to artificial concatenated sequences can increase the quality of the dataset. Users may modify the different filter parameters according to their own preferences.
TagCleaner is a publicly available web application that is able to automatically detect and efficiently remove tag sequences from metagenomic datasets. It is easily configurable and provides a user-friendly interface. The interactive web interface facilitates export functionality for subsequent data processing, and is available at http://edwards.sdsu.edu/tagcleaner.
使用基于引物的方法对宏基因组进行预扩增后,需要从数据集中去除额外的标签序列。由于测序的限制,测序reads 可能包含缺失或插入,并且引物序列可能包含模糊碱基。此外,标签序列可能不可用或报告不正确。由于下游序列污染引入的不准确的潜在可能性,使用可靠的工具对序列数据进行预处理非常重要。
TagCleaner 是一个开发的网络应用程序,用于自动识别和去除已知或未知的标签序列,同时允许数据集插入和缺失。TagCleaner 旨在过滤修剪后的 reads 中的重复、短 reads 和高模糊序列率的 reads。额外的片段到片段拼接的筛选和拆分,这些拼接产生了人为拼接序列,可以提高数据集的质量。用户可以根据自己的喜好修改不同的过滤参数。
TagCleaner 是一个公开可用的网络应用程序,能够自动检测和从宏基因组数据集中有效地去除标签序列。它易于配置,并提供了用户友好的界面。交互式网络界面方便了后续数据处理的导出功能,可在 http://edwards.sdsu.edu/tagcleaner 上获取。