Srivastava Sudeep, Chen Liang
Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Nucleic Acids Res. 2010 Sep;38(17):e170. doi: 10.1093/nar/gkq670. Epub 2010 Jul 29.
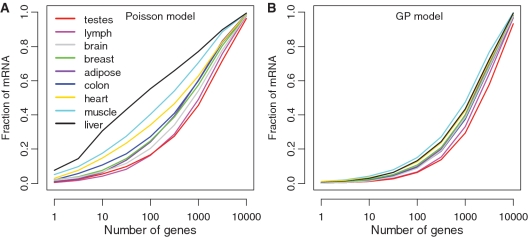
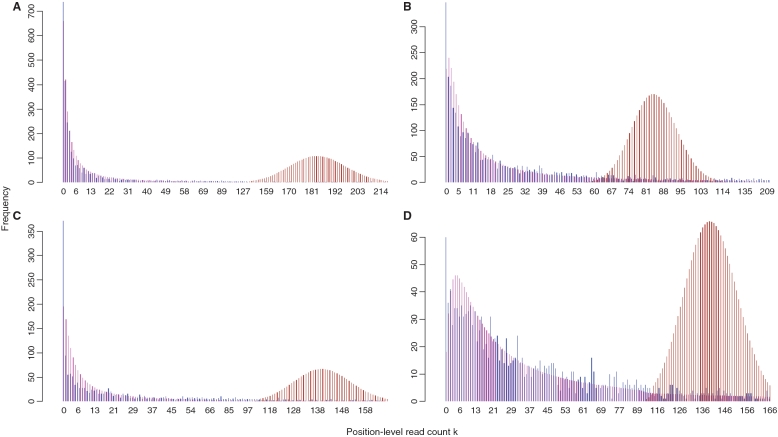
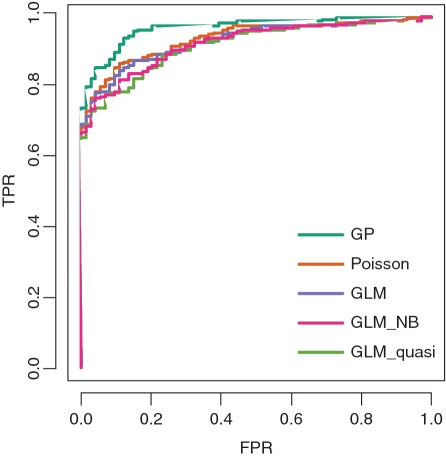
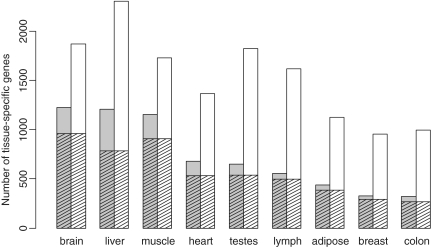
Deep sequencing of RNAs (RNA-seq) has been a useful tool to characterize and quantify transcriptomes. However, there are significant challenges in the analysis of RNA-seq data, such as how to separate signals from sequencing bias and how to perform reasonable normalization. Here, we focus on a fundamental question in RNA-seq analysis: the distribution of the position-level read counts. Specifically, we propose a two-parameter generalized Poisson (GP) model to the position-level read counts. We show that the GP model fits the data much better than the traditional Poisson model. Based on the GP model, we can better estimate gene or exon expression, perform a more reasonable normalization across different samples, and improve the identification of differentially expressed genes and the identification of differentially spliced exons. The usefulness of the GP model is demonstrated by applications to multiple RNA-seq data sets.
RNA的深度测序(RNA-seq)已成为表征和定量转录组的有用工具。然而,RNA-seq数据分析存在重大挑战,例如如何从测序偏差中分离信号以及如何进行合理的标准化。在此,我们关注RNA-seq分析中的一个基本问题:位置水平读取计数的分布。具体而言,我们针对位置水平读取计数提出了一种双参数广义泊松(GP)模型。我们表明,GP模型比传统泊松模型对数据的拟合要好得多。基于GP模型,我们可以更好地估计基因或外显子表达,在不同样本间进行更合理的标准化,并改善差异表达基因的识别以及差异剪接外显子的识别。通过将GP模型应用于多个RNA-seq数据集,证明了该模型的实用性。