Department of Microbiology and Immunology, the University of Rochester School of Medicine and Dentistry, Rochester, New York, United States of America.
PLoS One. 2010 Jul 29;5(7):e11875. doi: 10.1371/journal.pone.0011875.
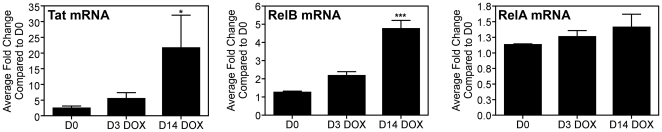
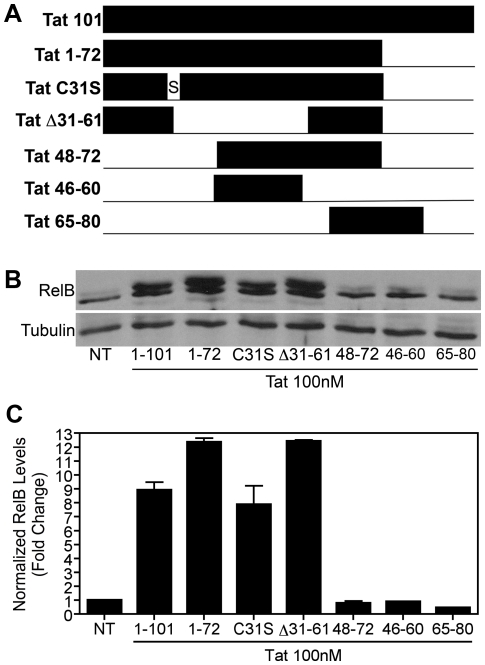
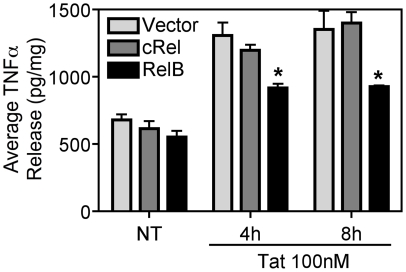
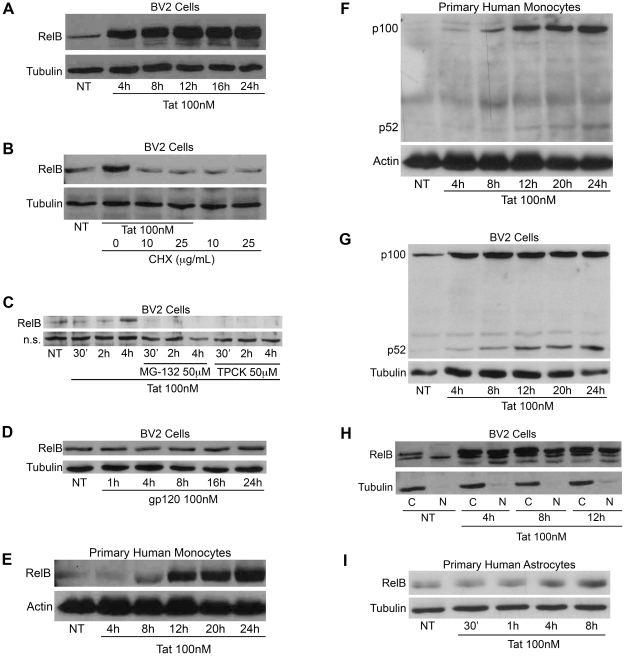
Human Immunodeficiency Virus-1 (HIV-1)-associated neurocognitive disorder (HAND) is likely neuroinflammatory in origin, believed to be triggered by inflammatory and oxidative stress responses to cytokines and HIV protein gene products such as the HIV transactivator of transcription (Tat). Here we demonstrate increased messenger RNA for nuclear factor-kappa B (NF-kappaB) family member, transcription factor RelB, in the brain of doxycycline-induced Tat transgenic mice, and increased RelB synthesis in Tat-exposed microglial cells. Since genetic ablation of RelB in mice leads to multi-organ inflammation, we hypothesized that Tat-induced, newly synthesized RelB inhibits cytokine production by microglial cells, possibly through the formation of transcriptionally inactive RelB/RelA complexes. Indeed, tumor necrosis factor-alpha (TNFalpha) production in monocytes isolated from RelB deficient mice was significantly higher than in monocytes isolated from RelB expressing controls. Moreover, RelB overexpression in microglial cells inhibited Tat-induced TNFalpha synthesis in a manner that involved transcriptional repression of the TNFalpha promoter, and increased phosphorylation of RelA at serine 276, a prerequisite for increased RelB/RelA protein interactions. The Rel-homology-domain within RelB was necessary for this interaction. Overexpression of RelA itself, in turn, significantly increased TNFalpha promoter activity, an effect that was completely blocked by RelB overexpression. We conclude that RelB regulates TNFalpha cytokine synthesis by competitive interference binding with RelA, which leads to downregulation of TNFalpha production. Moreover, because Tat activates both RelB and TNFalpha in microglia, and because Tat induces inflammatory TNFalpha synthesis via NF-kappaB, we posit that RelB serves as a cryoprotective, anti-inflammatory, counter-regulatory mechanism for pathogenic NF-kappaB activation. These findings identify a novel regulatory pathway for controlling HIV-induced microglial activation and cytokine production that may have important therapeutic implications for the management of HAND.
人类免疫缺陷病毒 1 型(HIV-1)相关的神经认知障碍(HAND)可能源于神经炎症,据信是由细胞因子和 HIV 蛋白基因产物(如 HIV 转录激活物(Tat))引起的炎症和氧化应激反应引发的。在这里,我们证明了在强力霉素诱导的 Tat 转基因小鼠的大脑中,核因子-κB(NF-κB)家族成员转录因子 RelB 的信使 RNA 增加,并且在暴露于 Tat 的小胶质细胞中合成了更多的 RelB。由于在小鼠中基因敲除 RelB 会导致多器官炎症,因此我们假设 Tat 诱导的新合成的 RelB 通过形成转录无活性的 RelB/RelA 复合物来抑制小胶质细胞中细胞因子的产生。事实上,从小鼠中分离的单核细胞中 TNFα 的产生在 RelB 缺陷型小鼠中明显高于从表达控制的 RelB 表达的单核细胞中。此外,在小胶质细胞中过表达 RelB 以涉及 TNFα 启动子转录抑制的方式抑制 Tat 诱导的 TNFα 合成,并增加 RelA 在丝氨酸 276 处的磷酸化,这是增加 RelB/RelA 蛋白相互作用的前提。RelB 中的 Rel 同源结构域对于这种相互作用是必需的。反过来,RelA 本身的过表达会显著增加 TNFα 启动子活性,而 RelB 过表达完全阻断了这种效应。我们得出的结论是,RelB 通过与 RelA 的竞争性干扰结合来调节 TNFα 细胞因子的合成,从而导致 TNFα 产生的下调。此外,由于 Tat 在小胶质细胞中激活 RelB 和 TNFα,并且由于 Tat 通过 NF-κB 诱导炎症性 TNFα 合成,我们假设 RelB 作为一种保护、抗炎、抗炎症性 NF-κB 激活的代偿性机制。这些发现确定了控制 HIV 诱导的小胶质细胞激活和细胞因子产生的新调节途径,这可能对 HAND 的管理具有重要的治疗意义。