School of Computing Science, Simon Fraser University, Burnaby, Canada.
Bioinformatics. 2010 Sep 15;26(18):i625-31. doi: 10.1093/bioinformatics/btq393.
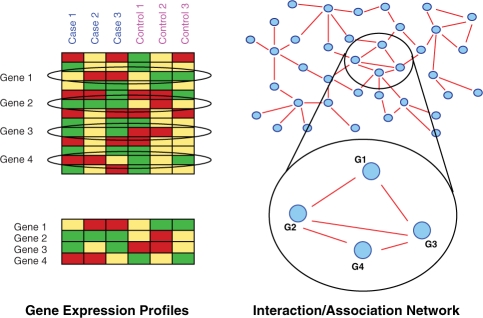
Recent genomic studies have confirmed that cancer is of utmost phenotypical complexity, varying greatly in terms of subtypes and evolutionary stages. When classifying cancer tissue samples, subnetwork marker approaches have proven to be superior over single gene marker approaches, most importantly in cross-platform evaluation schemes. However, prior subnetwork-based approaches do not explicitly address the great phenotypical complexity of cancer.
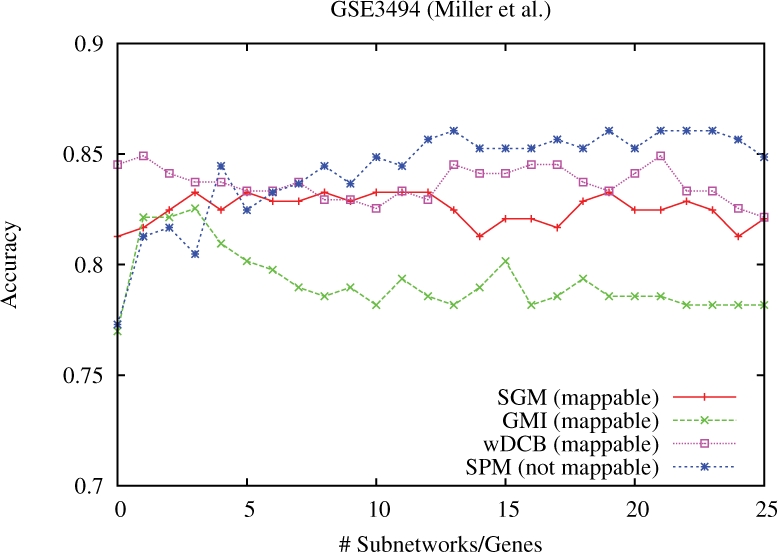
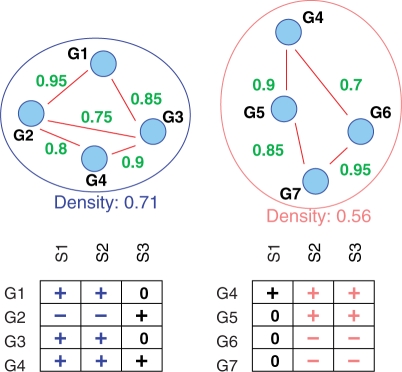
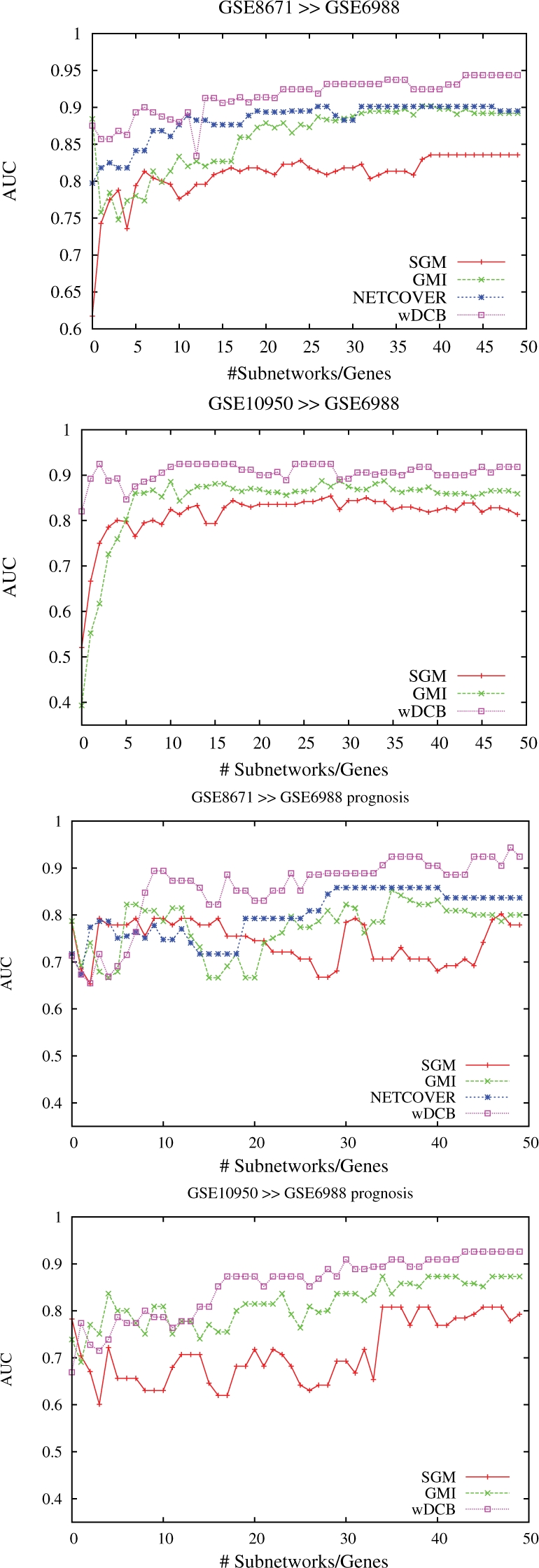
We explicitly address this and employ density-constrained biclustering to compute subnetwork markers, which reflect pathways being dysregulated in many, but not necessarily all samples under consideration. In breast cancer we achieve substantial improvements over all cross-platform applicable approaches when predicting TP53 mutation status in a well-established non-cross-platform setting. In colon cancer, we raise prediction accuracy in the most difficult instances from 87% to 93% for cancer versus non-cancer and from 83% to (astonishing) 92%, for with versus without liver metastasis, in well-established cross-platform evaluation schemes.
Software is available on request.
最近的基因组研究证实,癌症在表型上具有极高的复杂性,在亚型和进化阶段上有很大的差异。在对癌症组织样本进行分类时,子网标记方法已被证明优于单个基因标记方法,尤其是在跨平台评估方案中。然而,以前基于子网的方法并没有明确解决癌症的巨大表型复杂性。
我们明确地解决了这个问题,并采用密度约束的二分聚类来计算子网标记,这些标记反映了在许多但不一定是所有考虑中的样本中失调的途径。在乳腺癌中,我们在一个经过良好验证的非跨平台设置中,在预测 TP53 突变状态方面,与所有跨平台适用的方法相比,取得了实质性的改进。在结肠癌中,我们在最困难的情况下,将癌症与非癌症之间的预测准确率从 87%提高到 93%,将有肝转移和无肝转移之间的预测准确率从 83%提高到(惊人的)92%,在经过良好验证的跨平台评估方案中。
软件可根据要求提供。