Committee on Higher Degrees in Biophysics, Harvard University, Cambridge, Massachusetts 02139-4307, United States.
J Am Chem Soc. 2010 Oct 27;132(42):14919-27. doi: 10.1021/ja105832g.
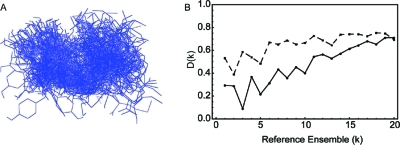
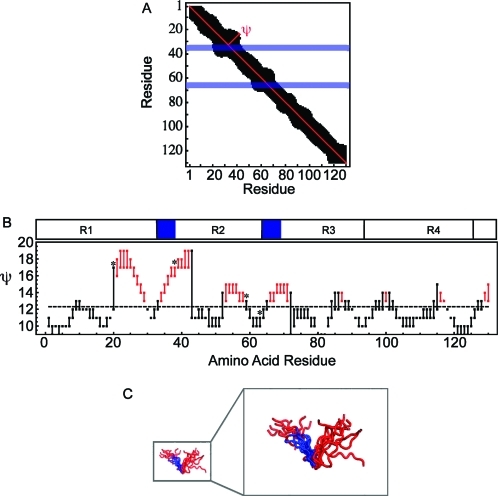
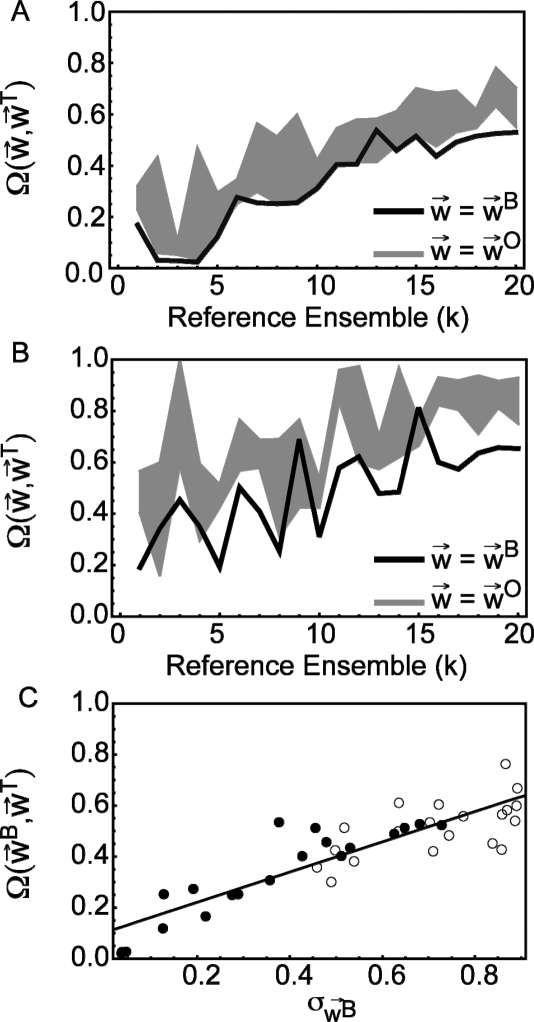
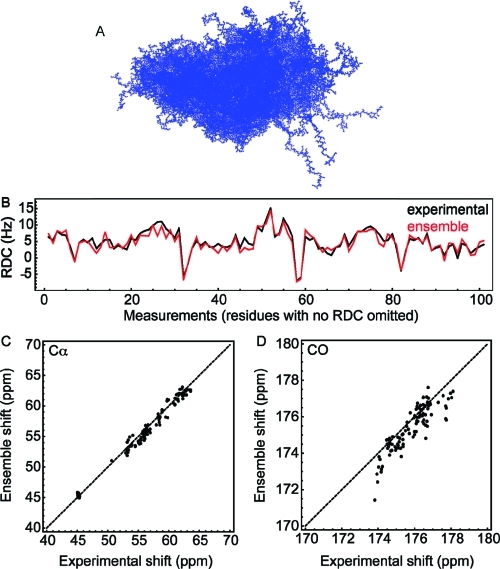
The characterization of intrinsically disordered proteins is challenging because accurate models of these systems require a description of both their thermally accessible conformers and the associated relative stabilities or weights. These structures and weights are typically chosen such that calculated ensemble averages agree with some set of prespecified experimental measurements; however, the large number of degrees of freedom in these systems typically leads to multiple conformational ensembles that are degenerate with respect to any given set of experimental observables. In this work we demonstrate that estimates of the relative stabilities of conformers within an ensemble are often incorrect when one does not account for the underlying uncertainty in the estimates themselves. Therefore, we present a method for modeling the conformational properties of disordered proteins that estimates the uncertainty in the weights of each conformer. The Bayesian weighting (BW) formalism incorporates information from both experimental data and theoretical predictions to calculate a probability density over all possible ways of weighting the conformers in the ensemble. This probability density is then used to estimate the values of the weights. A unique and powerful feature of the approach is that it provides a built-in error measure that allows one to assess the accuracy of the ensemble. We validate the approach using reference ensembles constructed from the five-residue peptide met-enkephalin and then apply the BW method to construct an ensemble of the K18 isoform of the tau protein. Using this ensemble, we indentify a specific pattern of long-range contacts in K18 that correlates with the known aggregation properties of the sequence.
无定形蛋白质的特性描述具有挑战性,因为这些系统的准确模型需要描述其热可及构象和相关的相对稳定性或权重。这些结构和权重通常是这样选择的,即计算的总体平均值与一组预定义的实验测量值一致;然而,这些系统中的自由度数量通常导致与任何给定的实验可观察量集相关的多个构象总体。在这项工作中,我们证明了当不考虑估计本身的潜在不确定性时,对总体中构象相对稳定性的估计往往是不正确的。因此,我们提出了一种用于模拟无定形蛋白质构象特性的方法,该方法估计了总体中每个构象的权重的不确定性。贝叶斯加权(BW)形式主义结合了实验数据和理论预测的信息,以计算总体中加权构象的所有可能方式的概率密度。然后,使用该概率密度来估计权重的值。该方法的一个独特而强大的特点是,它提供了一种内置的误差度量,允许人们评估总体的准确性。我们使用由五个残基组成的肽 met-enkephalin 构建的参考总体来验证该方法,然后应用 BW 方法来构建 tau 蛋白 K18 异构体的总体。使用这个总体,我们确定了 K18 中长程接触的特定模式,该模式与序列的已知聚集特性相关。