Department of Environmental Sciences & Engineering, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
BMC Genomics. 2010 Oct 18;11:574. doi: 10.1186/1471-2164-11-574.
Analysis of microarray experiments often involves testing for the overrepresentation of pre-defined sets of genes among lists of genes deemed individually significant. Most popular gene set testing methods assume the independence of genes within each set, an assumption that is seriously violated, as extensive correlation between genes is a well-documented phenomenon.
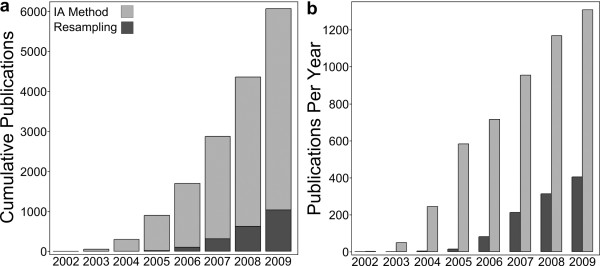
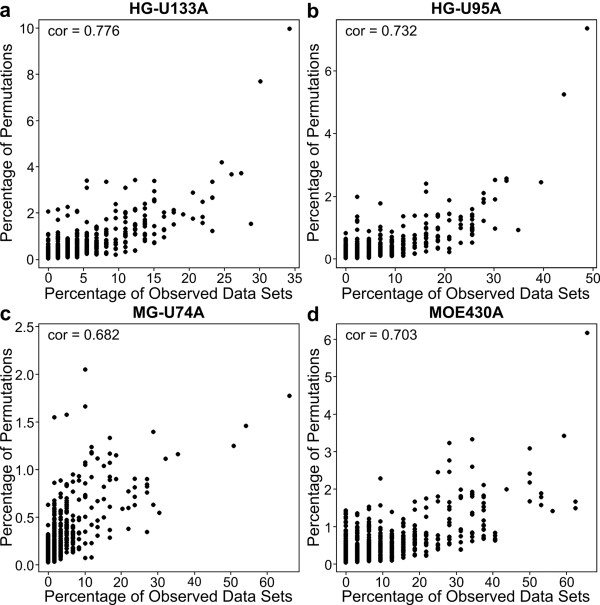
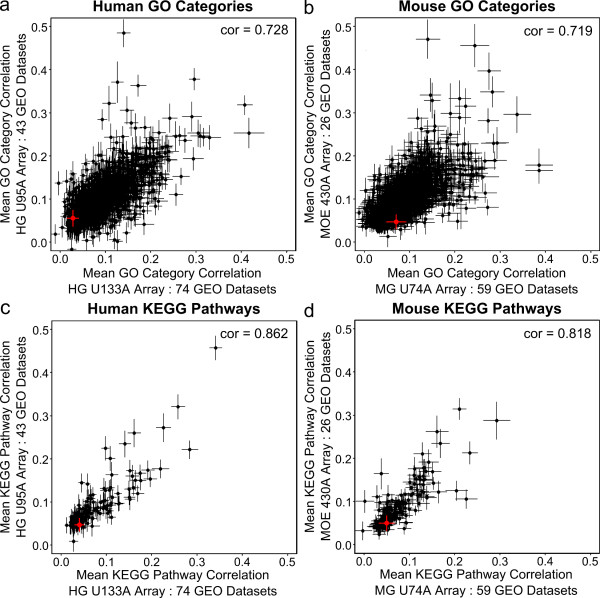
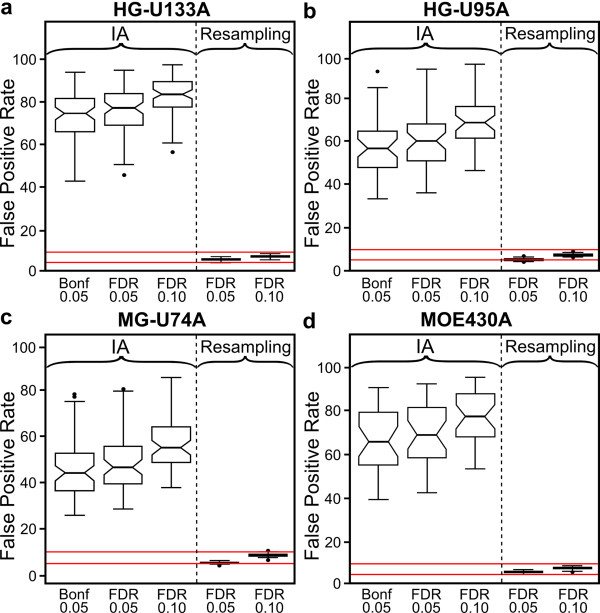
We conducted a meta-analysis of over 200 datasets from the Gene Expression Omnibus in order to demonstrate the practical impact of strong gene correlation patterns that are highly consistent across experiments. We show that a common independence assumption-based gene set testing procedure produces very high false positive rates when applied to data sets for which treatment groups have been randomized, and that gene sets with high internal correlation are more likely to be declared significant. A reanalysis of the same datasets using an array resampling approach properly controls false positive rates, leading to more parsimonious and high-confidence gene set findings, which should facilitate pathway-based interpretation of the microarray data.
These findings call into question many of the gene set testing results in the literature and argue strongly for the adoption of resampling based gene set testing criteria in the peer reviewed biomedical literature.
微阵列实验的分析通常涉及测试在被认为单独显著的基因列表中预定义的基因集的过表达。大多数流行的基因集测试方法假设每个集中的基因是独立的,这一假设严重违反了,因为基因之间存在广泛的相关性是一个有据可查的现象。
我们对来自基因表达综合数据库的 200 多个数据集进行了荟萃分析,以证明在实验中高度一致的强基因相关模式的实际影响。我们表明,当应用于已随机分组的数据集时,基于常见独立性假设的基因集测试程序会产生非常高的假阳性率,并且具有高内部相关性的基因集更有可能被宣布为显著。使用数组重采样方法对相同数据集进行重新分析可以正确控制假阳性率,从而得出更简洁、更可信的基因集发现,这将有助于基于途径的微阵列数据分析解释。
这些发现对文献中的许多基因集测试结果提出了质疑,并强烈呼吁在同行评审的生物医学文献中采用基于重采样的基因集测试标准。